CHAPTER 15 Elevated Cardiac Troponin in the Absence of Acute Coronary Syndromes
Mechanism, Significance, and Prognosis
Diagnostic criteria used for defining MI have changed significantly in recent years. The previously used World Health Organization definition required the presence of at least two criteria (typical clinical presentation, ischemic changes on ECG, and elevated total creatine kinase (CK) or its MB isoenzyme [CK-MB]) to diagnose an MI properly.1 An individual with absent biomarker elevation could have been diagnosed with an MI in the appropriate clinical setting and if ECG changes existed. With the advent of more sensitive myocardial biomarkers—the cardiac troponins—new definitions have since been proposed. In 2000, a joint committee of the European Society of Cardiology/American College of Cardiology (ESC/ACC) emphasized the presence of troponin elevation and proposed a new definition for acute MI, stating that “any amount of myocardial damage” (as evidenced by the troponin level) regardless of the magnitude, “implies an impaired clinical outcome,” and should be characterized as an MI.2 The 2002 American College of Cardiology/American Heart Association practice guidelines for the management of patients with unstable angina and non–ST segment elevation MI also reflected this view.3
Execution of these new guidelines into clinical practice has significantly increased the frequency of MI diagnoses. Kontos and colleagues4 reported that among 2181 patients presenting to the emergency department with chest pain, MI diagnoses based on cardiac troponin level increased 195% compared with diagnoses made according to CK-MB criteria. Troponin testing is now also being used as a screening tool in patients with a low pretest probability of thrombotic coronary artery disease. Because of its high sensitivity for detecting even minimal myocardial cell necrosis, troponin may become “positive” even in the absence of thrombotic obstruction of a coronary artery. In many instances, these troponin elevations may occur due to demand ischemia, with minor epicardial coronary obstruction (or even without significant coronary artery disease), rather than an acute thrombotic coronary occlusion.5 One retrospective study comprising 166 patients presenting to the emergency department with elevated troponin showed that only 61% of the patients had significant coronary stenosis as per coronary angiography.6 Ng and colleagues7 showed that of 112 patients presenting to the emergency department with increased troponin I levels, 45% had a final diagnosis other than ACS.
Biochemistry and Assays of Troponins
Cardiac troponins are regulatory proteins found on the contractile apparatus of striated muscle that control the calcium-mediated interaction of actin and myosin.8,9 They form complexes with actin and tropomyosin on the thin filament of the contractile apparatus and regulate contraction by initiating or inhibiting the sliding of thin filaments over thick filaments. The troponin complex consists of three subunits (Fig. 15-1): troponin C, which binds to calcium ions and regulates activation of the thin filaments during contraction; troponin T, which binds to tropomyosin and facilitates contraction; and troponin I, which binds to actin and inhibits actin-myosin interactions in the absence of calcium.8,10,11
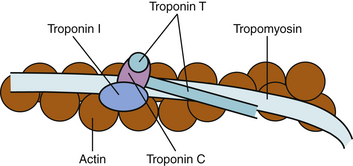
The amino acid sequences of the skeletal and cardiac isoforms of cardiac troponin T and troponin I are dissimilar. Monoclonal antibodies to both of these cardiac troponins have been developed with no cross-reactivity with troponins from skeletal muscle.3 In contrast, cardiac and skeletal muscle share troponin C isoforms; no clinical assay has been developed to test for this protein because of its lack of cardiac specificity.
Current troponin assays are very sensitive, detecting even small amounts of myocardial necrosis. The ESC/ACC Committee defined MI as a troponin level greater than the upper reference limit of the healthy population—that is, greater than the 99th percentile of the reference control group with a coefficient of variation of 10% or less. Because very few assays being used meet this standard, it has been proposed that a troponin elevation greater than the 10% coefficient of variation be indicative of myocardial injury.12 Although this reference limit may slightly reduce the sensitivity for MI according to the biochemical diagnostic criterion, it allows providers to avoid being faced with minor troponin elevations that may simply be secondary to analytic noise. With a wide variety of commercial assays being used, each institution’s laboratory must first confirm the reference range in their particular population before defining their cutoff for a “significant” troponin elevation.
Troponin and Myocardial Damage
Approximately 7% of troponin T and 3% to 5% of troponin I is normally found free in the myocyte cytoplasm, whereas most is structurally bound in the contractile apparatus of the muscle fiber.13 The release of troponin from the myocyte to the blood can be due to reversible or irreversible cell damage.8 A biphasic increase in serum troponin occurs during myocardial ischemia, corresponding to the early release of free cytoplasmic proteins, followed by a gradual release of myofibril-bound cytosolic complexes as the myocytes are irreversibly damaged, and the cell membrane degrades.8
Speculations have been made regarding the mechanism of this early troponin release. Observations of patients with unstable angina have shown only transient troponin elevations, with values returning to baseline within a few hours.14 This pattern of early troponin increase may not necessarily be associated with irreversible myocardial necrosis. It is conceivable that myocardial troponin can also be released in the setting of increased membrane permeability. It is thought that myocardial depressive factors (released in the setting of sepsis and other inflammatory states) cause degradation of free troponin in situ to lower molecular weight fragments.15 With increased membrane permeability, the smaller troponin fragments could be released into the systemic circulation. In this setting, myocyte damage may not be permanent, and cell necrosis does not occur. This notion is also supported by the clinical observation that myocardial depression during sepsis is a fully reversible process in most surviving patients.16
Nonthrombotic Mechanisms of Troponin Elevation
Troponin elevation can occur in the absence of ACS. Frequent conditions causing troponin elevation include hypovolemia, sepsis, heart failure, atrial fibrillation, myocarditis, myocardial contusion, pulmonary embolus, and renal failure (Table 15-1).
Table 15–1 Nonthrombotic Causes and Presumed Mechanism for Elevated Cardiac Troponin Level
| Diagnosis | Mechanism |
|---|---|
| Demand Ischemia | |
| Sepsis/systemic inflammatory response syndrome | Myocardial depression/supply-demand mismatch |
| Hypotension | Decreased perfusion pressure |
| Hypovolemia | Decreased filling pressure/output |
| Supraventricular tachycardia/atrial fibrillation | Supply-demand mismatch |
| Left ventricular hypertrophy | Subendocardial ischemia |
| Myocardial Ischemia | |
| Coronary vasospasm | Prolonged ischemia with myonecrosis |
| Intracranial hemorrhage or stroke | Imbalance of autonomic nervous system |
| Ingestion of sympathomimetic agents | Direct adrenergic effects |
| Direct Myocardial Damage | |
| Cardiac contusion | Traumatic |
| Direct current cardioversion | Traumatic |
| Cardiac infiltrative disorders | Myocyte compression |
| Chemotherapy | Cardiotoxicity |
| Myocarditis | Inflammatory |
| Pericarditis | Inflammatory |
| Cardiac transplantation | Inflammatory/immune-mediated |
| Myocardial Strain | |
| Congestive heart failure | Myocardial wall stretch |
| Pulmonary embolism | Right ventricular stretch |
| Pulmonary hypertension or emphysema | Right ventricular stretch |
| Strenuous exercise | Ventricular stretch |
| Chronic renal insufficiency | Unknown |
From Jeremias A, Gibson CM: Narrative review: alternative causes for elevated cardiac troponin levels when acute coronary syndromes are excluded. Ann Intern Med 2005;142:787.
Demand Ischemia
The term demand ischemia refers to inadequate myocardial oxygen supply in the setting of increased demand and in the absence of a thrombotic coronary artery occlusion. Myocardial oxygen demand increases in several clinical conditions, including sepsis or the systemic inflammatory response syndrome,17–19 hypotension or hypovolemia,20 and atrial fibrillation or other tachyarrhythmias.21,22 These disease states can predispose to tachycardia and various loading conditions on the heart. Tachycardia can augment myocardial oxygen demand while decreasing myocardial oxygen supply predominantly by reducing the time of diastole, the time period in which much of myocardial perfusion occurs. Also, systemic inflammatory processes, including sepsis, can result in higher oxygen consumption, decreased perfusion pressure, myocardial depression, and reduced delivery of oxygen to the heart, resulting in release of troponin into the systemic circulation.18
Elevated troponin is a common finding in critically ill patients and is associated with a significant increase in mortality.17 Ammann and colleagues18 reported that in 20 intensive care unit (ICU) patients with sepsis, 85% had elevated troponin levels, 59% of which had no evidence of significant coronary artery disease. A more recent study evaluating 58 ICU patients without ACS found that of the 55% that had elevated troponins, significant coronary artery disease was excluded in 72% of these patients. Levels of C-reactive protein, tumor necrosis factor-α, and interleukin-6 were markedly higher among troponin-positive patients.19 Mortality was also fourfold higher in this group. Troponin elevation is common in ICU patients without thrombotic ACS. No definitive causal relationship has yet been shown; however, it has been proposed that the inflammatory mediators involved in systemic inflammatory states may contribute to the myocardial oxygen demand-supply mismatch. In addition, whether any cardiovascular work-up or intervention could improve the mortality risk of these patients remains unclear.
Tachycardia and several tachyarrhythmias are other potential causes of demand ischemia in the absence of thrombotic coronary artery disease. Bakshi and colleagues21 showed that among 21 patients with elevated troponin levels but a normal coronary angiogram, tachycardia was the cause in 28%, strenuous exercise was the cause in 10%, pericarditis was the cause in 10%, and congestive heart failure was the cause in 5%; 47% of patients had no identifiable trigger.21 Similarly, Zellweger and associates22 described four patients with supraventricular tachycardia with increased levels of troponin but no evidence of epicardial coronary stenoses. It is evident from these reports that troponin release can be a result of tachycardia alone, and can be released in the absence of coronary artery disease, inflammatory mediators, and myocardial depressive factors.
Elevated cardiac troponin has also been noted in the setting of left ventricular hypertrophy. Hamwi and colleagues23 reported that in 74 patients without any clinical evidence of active myocardial ischemia, patients in the upper tertile of left ventricular mass had increased troponin levels compared with patients in the lowest tertile. The increased left ventricular mass necessitates a greater myocardial oxygen demand and may induce occult subendocardial ischemia. Also, flow reserve is decreased secondary to the remodeling of coronary microcirculation. Similarly, elevated troponins in the setting of aortic valve disease are thought to be associated with greater left ventricular mass and higher pulmonary artery systolic pressures.24
Myocardial Ischemia with Dynamic Coronary Artery Obstruction
Myocardial ischemia, in the absence of obstructive ACS, can also be caused by vasospasm, also termed Prinzmetal angina. Wang and coworkers25 evaluated 93 patients with suspected myocardial ischemia and a final diagnosis of nonobstructive coronary artery disease, and found that 25% of these patients had increased troponins. Ergonovine provocation testing subsequently revealed vasoconstriction in 74% of these patients, suggesting that prolonged ischemia can be induced by coronary vasospasm.
Related posts:
 Evolution of the Coronary Care Unit: Past, Present, and Future
Evolution of the Coronary Care Unit: Past, Present, and Future
 Hemodynamically Unstable Presentations of Congenital Heart Disease in Adults
Hemodynamically Unstable Presentations of Congenital Heart Disease in Adults
 Vascular Complications after Percutaneous Coronary Intervention
Vascular Complications after Percutaneous Coronary Intervention
 Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
Diagnosis and Treatment of Complications of Coronary and Valvular Interventions
 Pharmacologic Interactions in the CICU
Pharmacologic Interactions in the CICU
 Acute Aortic Syndromes: Diagnosis and Management
Acute Aortic Syndromes: Diagnosis and Management
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


