(1)
Division of Pulmonary and Critical Care Medicine, Eastern Virginia Medical School, Norfolk, VA, USA
Keywords
Electrolyte disturbanceHyponatremiaHypernatremiaHypokalemiaHyperkalemiaHypocalcemiaHypercalcemiaHypophosphatemiaHypomagnesemiaSodium and Water
Rules of the Game
Sodium balance determines volume status.
Water balance determines tonicity i.e. Na+ concentration.
Volume Overload = increased total body sodium (regardless of serum Na concentration)
Euvolemia = normal total body sodium (regardless of serum Na concentration)
Volume Depletion = decreased total body sodium (regardless of serum Na concentration)
Hyponatremia = relative water excess
Hypernatremia = relative water deficit
In volume depleted patients, volume should be corrected prior to correction of tonicity; i.e. patients should initially be volume resuscitated with Ringers Lactate regardless of serum sodium concentration (see Chap. 9).
Hyponatremic dehydration; volume replacement with 0.9 % NaCl
Hypertonic dehydration: volume replacement with Ringers Lactate then change to 0.45 % NaCl.
Hyponatremia
Hyponatremia defined as a serum sodium <135 mEq/L, is the most common electrolyte abnormality encountered worldwide and is an independent risk factor for higher morbidity and mortality rates [1, 2]. The meta-analysis by Corona et al. demonstrated that the hyponatremia-related risk of overall mortality was inversely correlated with serum sodium concentration [2]. Symptoms related to hyponatremia have been traditionally associated with severe hyponatremia and acute reductions in serum sodium, but there is growing awareness that even mild hyponatremia is associated with mental dysfunction, unsteady gait and osteoporosis [2– 4].
The traditional diagnostic approach and management of hyponatremia is based on an assessment of the patient’s volume status; i.e. fluid overloaded, euvolemia or dehydration (see Fig. 40.1). This diagnostic workup includes measurement of urine and serum electrolytes and osmolarity, thyroid function tests, lipid profile and serum cortisol (and possibly ACTH stimulation test). The problem with this approach is that the clinical assessment of volume status is notoriously unreliable and that in many patients it is difficult to determine their volume status with any degree of accuracy (see Chap. 9). This distinction is of utmost importance in distinguishing SIADH from cerebral/renal salt wasting syndrome [3]. The only accurate method to determine volume status is by direct measurement using radiolabelled albumin or RBC. When the true circulating blood volume is measured many patients with SIADH are shown to be volume depleted and likely to have renal salt wasting [3].
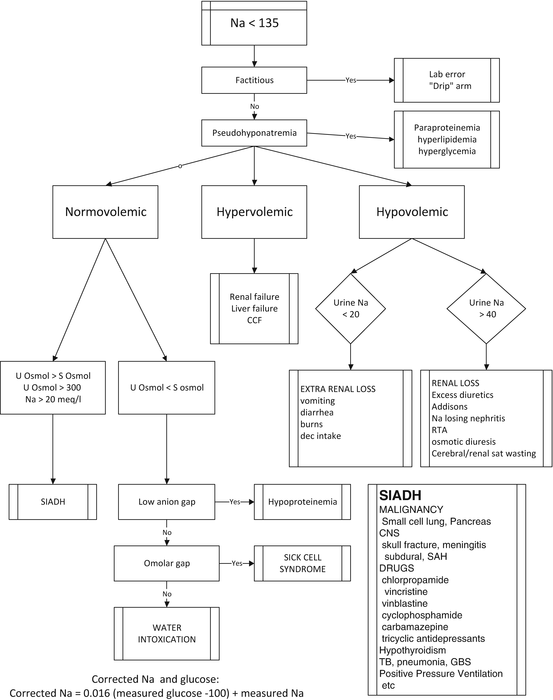
Fig. 40.1
Traditional approach to hyponatremia based on the patient’s volume status
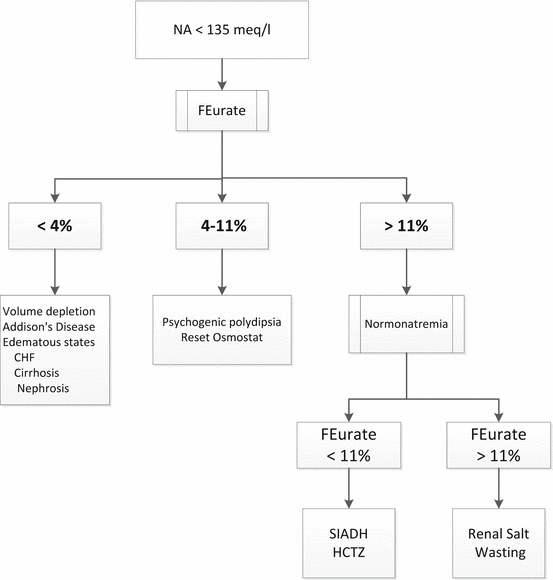
Maesaka et al. have proposed a new approach to hyponatremia based on the fractional excretion of urate (FEurate) (see Fig. 40.2) [3]. FEurate is normally 4– 11 %. FEurate is decreased in volume depleted states and increased in SIADH and RSW. In SIADH, correction of hyponatremia will normalize FEurate while it remains persistently increased in RSW. The value of determining FEurate in hyponatremic conditions has been further amplified by a normal FEurate in patients with Reset Osmostat [5].
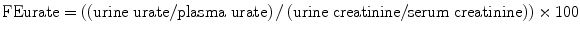
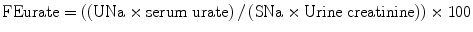 It should be noted that both SIADH and cerebral/renal salt wasting syndromes are associated with intracranial diseases, have normal renal, thyroid and adrenal function and have similar electrolyte abnormalities (see below) [3].
It should be noted that both SIADH and cerebral/renal salt wasting syndromes are associated with intracranial diseases, have normal renal, thyroid and adrenal function and have similar electrolyte abnormalities (see below) [3].
Fig. 40.2
Revised approach to hyponatremia based on FEurate
Findings common to both SIADH and RSW
Hyponatremia
Concentrated urine
Urine Na >40 mEq/L
Hyperuricemia with increased FEurate
Most patients with hyponatremia are “asymptomatic” and have a plasma sodium concentration above 120 mEq/L. In these patients there is no urgency in correcting the serum sodium concentration and treatment should occur over a number of days. Treatment typically consists of isotonic saline if the patient has true volume depletion or water restriction in the syndrome of inappropriate ADH secretion (SIADH). More aggressive therapy is indicated in those patients who have symptomatic or severe hyponatremia (plasma sodium concentration below 110 mEq/L). In this setting, hypertonic saline can be given initially to raise the plasma sodium concentration (to ~120 mEq/L), although the rate at which this occurs must be carefully monitored to minimize the risk of central demyelinating lesions (see below). Hyponatremia in the setting of effective intravascular volume depletion and edema (congestive cardiac failure and cirrhosis) results from release of ADH in response to “hypovolemia” and carries a poor prognosis. Such patients require volume expansion with albumin and attempts to correct the underlying hemodynamic derangement. The role of ADH antagonists in these patients is unclear [6, 7].
The treatment of hyponatremia requires either the addition of sodium or removal of water. When excess water (e.g. SIADH) rather than sodium deficit is the main mechanism of hyponatremia, removal of water alone is indicated. Net water removal can be achieved by the removal of sodium and water with a loop diuretic administered simultaneously with sodium in the form of normal saline or hypertonic saline. The administration of hypertonic saline alone will also induce sodium and water diuresis, resulting in net loss of water, but the urine concentration is less predictable in this situation. SIADH is usually treated by fluid restriction. Fluid restriction is frequently problematic in critically ill patients, particularly in neurosurgical patients in whom maintenance of a high intervascular volume is necessary to maintain cerebral perfusion. In these patients the use of a ADH receptor antagonist may be preferable (since the problem is water overload and no sodium deficiency). However, as alluded to above it may be exceedingly difficult to distinguish SIADH for CSW syndrome. In these patients it may be preferable to treat the patient with hypertonic saline, monitoring the change in serum sodium (carefully) and FEurate. In those patients who clearly have SIADH (very low BUN and features of hemodilution) an ADH receptor antagonist may be preferable whereas in patients who clearly have CSW (increased BUN and features of volume depletion) treatment with normal saline is preferable.
Patients with SIADH are euvolemic and have traditionally been treated with volume restriction, demeclocycline, salt tablets, hypertonic saline and/or fludrocortisone [1, 8– 11]. These interventions are not particularly effective and are unpredictable. Furthermore, volume restriction may result in intravascular volume depletion and impaired cerebral perfusion. Arginine vasopressin (AVP) binds to vasopressin-2 (V2) receptors located on the epithelial cells of the renal collecting ducts increasing apical cell membrane water permeability, which stimulates water reabsorption, thereby decreasing serum sodium [12]. Thus, a potentially valuable pharmacologic approach for correcting dilutional hyponatremia is AVP-receptor antagonism to block this antidiuretic cascade. Conivaptan is a high-affinity non-peptide antagonist of AVP V2 and V1A receptors that produces aquaresis, the renal excretion of solute-free water that is electrolyte sparing. Conivaptan is available in an intravenous formation. Tolvaptan is as selective V2 receptor antagonist that is only available in an oral formulation. We have previously reported our experience with the use of a single dose of 20 mg conivaptan (infused over 30 min) in 32 neurocritical care patients with presumed SIADH [13]. The mean 24 h natremic response was 4.3 ± 2.6 mEq/L (range of 1– 13 mEq/L), with the greatest change occurring within the first 8 h. Eighteen (56 %) patients met the primary end-point of an increase in serum sodium of ≥4 mEq/L in 24 h. The change in serum sodium from 24 to 48 h was 0.5 ± 1.3 mEq/L. We noted no side effects or cases of phlebitis. These results are similar to those reported in the literature and allows for a controlled and predictable increase in the serum sodium concentration [12]. Murphy et al. reported that the maximal response to a single dose of 20 mg conivaptan was maximal at 12 h with the effect being sustained over 72 h [14]. In the study by Murphy et al. the increase in urine output was maximal at 2 h and returned to baseline at 12 h. Rapid and excessive correction of sodium has not been reported with conivaptan [15, 16]. The largest 24 h natremic response in our study was 13 mEq/L. Similar results were reported by the Conivaptan Study group [12]. Conivaptan may act optimally when given as bolus doses rather than as a continuous infusion. Infusion-site reactions and venous phlebitis occurred in up to 73 % of patients receiving a 20 mg/day infusion in company-sponsored trials [16]. Repeat dosing together with the use of normal saline (and a small dose of a loop diuretic) is suggested in those who respond poorly to conivaptan. In all patients receiving AVP antagonists and hypertonic saline the serum sodium must be very closely monitored and corrective action taken if the serum sodium is increasing to quickly (see below). Conivaptan (but not tolvaptan) has a number of important drug interactions. Conivaptan is a sensitive substrate of CYP3A. Conivaptan is a potent mechanism-based inhibitor of CYP3A and can increase the serum levels of midazolam, simvastatin and amlodipine [16].
The optimal rate of correction of hyponatremia varies with the clinical state of the patient. Acute symptomatic hyponatremia (less than 48 h) results in cerebral edema to due water movement into the brain. In contrast, chronic hyponatremia allows time for cerebral adaption to occur, resulting in the return of brain volume towards normal and usually causes no neurological symptoms. “Rapid” initial correction of hyponatremia is warranted in symptomatic patients, but overly rapid correction can be deleterious (osmotic demyelinating syndrome), particularly in patients with chronic hyponatremia [17]. The rate at which this correction should occur will depend on the presence of symptoms and the rate at which the disorder occurred. Alcoholic patients are particularly prone to cerebral demyelinosis. A recently convened panel of experts recommended that, based on the results of cohort studies and literature reviews, the optimal rate of sodium correction to avoid osmotic demyelination syndrome in patients with chronic hyponatremia is less than 12 mEq/L per 24 h and less than 18 mEq/L in 48 h [10]. In asymptomatic or mildly symptomatic patients the plasma sodium concentration should be raised at a maximal rate below 0.5 mEq/L per hour [18]. More rapid correction is indicated in patients with symptomatic hyponatremia who present with seizures or other severe neurologic manifestations; these findings are primarily due to cerebral edema induced by acute hyponatremia. In this setting, the plasma sodium concentration can be raised at an initial rate of 1.5– 2 mEq/L per hour for the first 3– 4 h, since the risk of persistent severe hyponatremia is greater than the possible danger of overly rapid correction. Despite the more rapid initial rate of correction with symptomatic hyponatremia, the total increase in the plasma sodium concentration over the first 24 h should not exceed 12 mEq/L, the same limit noted with asymptomatic patients.
The amount of Na+ required to raise the plasma Na+ concentration to a safe level (120 mEq/L) can be calculated from the following formula:
![$$ \mathrm{N}\mathrm{a}+\mathrm{deficit}=0.6\times \mathrm{lean}\;\mathrm{body}\;\mathrm{weight}\;\left(\mathrm{kg}\right)\times \left(120-\mathrm{plasma}\;\left[{\mathrm{Na}}^{+}\right]\right) $$](/wp-content/uploads/2016/10/A69851_3_En_40_Chapter_Equc.gif)
(Substitute 0.6 for 0.5 in women)
Since 3 % saline contains ~500 mEq/L, the total amount of this solution (in liters) required to increase the serum sodium to 120 mEq/L can be calculated as follows:
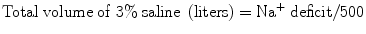 When 3 % saline is used the initial rate is usually between 50 and 100 mL/h; the serum sodium should be checked after 2 h and then 2 hourly until the sodium has stabilized. An alternative (and perhaps safer) approach in “euvolemic” hyponatremic patients is to use a 0.9 % saline infusion together with furosemide, to increase the free water loss.
When 3 % saline is used the initial rate is usually between 50 and 100 mL/h; the serum sodium should be checked after 2 h and then 2 hourly until the sodium has stabilized. An alternative (and perhaps safer) approach in “euvolemic” hyponatremic patients is to use a 0.9 % saline infusion together with furosemide, to increase the free water loss.
Hypernatremia
Hypernatremia is not uncommon in ICU patients primarily due to excessive resuscitation with 0.9 NaCl ([Na+] 154 mEq/L). Hypernatremia may also occur in dehydrated patients in whom thirst is impaired (e.g. bed ridden or altered mental status). The treatment of the former is free water (e.g. 200 mL water orally q 4– 6 h). In hypertonic dehydration volume status should be corrected first (Ringers Lactate) followed by correction of tonicity (0.45 % saline and/or free water). Rapid correction of hypernatremia can induce cerebral edema, seizures, permanent neurological damage and death. To minimize these risks the plasma Na+ concentration should be corrected slowly. The maximum rate at which the plasma Na+ should be corrected is 0.5 mEq/L/h or 12 mEq/L per day, a rate equivalent to that of hyponatremia. The water deficit can be calculated using the following formula:
![$$ \mathrm{Water}\;\mathrm{deficit}=0.5\times \left(\left(\left[{\mathrm{Na}}^{+}\right]/140\right)-1\right),\;\mathrm{substitute}\;0.5\;\mathrm{with}\;0.4\;\mathrm{in}\;\mathrm{women}. $$](/wp-content/uploads/2016/10/A69851_3_En_40_Chapter_Eque.gif)
Hypokalemia
Only a small fraction of the total body K+ is extracellular. Therefore serum K+ levels do not accurately reflect the total body K+. The degree of K+ deficit is dependent on the duration of the precipitating cause (time for equilibration) and the serum K+ level. In patients with chronic hypokalemia 1 mEq fall in serum K+ is approximately equal to a 200 mEq total body deficit. In critically ill ICU patients it is generally recommended to keep the serum [K+] ≥4.0 mEq/L. In patients with severe hypokalemia (K < 3.0 mEq/L) both PO and as well as IV potassium is recommended.
IV replacement therapy of KCl
No more than 20 meq/h should be given via a central line infusion
20 meq in 50 mL over 1 h
40 meq in 100 mL over 2 h
Peripheral line infusion
10 meq in 100 mL over 1 h
20 meq in 200 mL over 2 h
Hyperkalemia
Acute hyperkalemia is usually the result of renal failure. Hyperkalemia may also occur with overzealous potassium replacement and in patients receiving ACE inhibitors and potassium sparing diuretics. Factors such as the duration of hyperkalemia, the plasma Ca2+ concentration and the acid– base balance modify the toxicity of hyperkalemia. However, a K+ >6.5 mEq/L or hyperkalemia associated with ECG changes should be regarded as life-threatening requiring immediate treatment.
Clinical features usually occur when the K+ >6.5 mEq/L and include
Weakness
Paresthesia
Ileus
Paralysis
cardiac arrest
ECG changes include:
peaked T waves
flattened P
prolonged PR interval
widening of the QRS complex
sine wave leading to V. fibrillation or asystole
The rate of progression of the ECG changes is not predictable and patients may progress from minor ECG changes to dangerous conduction disturbances or arrhythmias within minutes. The ECG changes are exacerbated by coexisting hyponatremia, hypocalcemia, hypermagnesemia and acidosis.
Patients should be treated when the K+ is greater than 5.5 mEq/L; urgent treatment is required when the K+ >6.5 mEq/L. The goals of treatment are to:
protect the heart from the effects of K+ by antagonizing the effect on cardiac conduction (calcium)
to shift the K+ from the extracellular to intracellular compartment
reduce the total body potassium
Life threatening arrhythmia may occur at any time during therapy, hence, continuous ECG monitoring is required. Patients with a serum K+ >6.5 mEq/L and/or significant ECG changes should be treated immediately with calcium gluconate, followed by a glucose/insulin infusion and then an iron exchange resin.
Hypophosphatemia
Phosphorus is an essential component of phospholipid, nucleic-acids and plays an essential role in energy metabolism. Only about 1 % of the total body phosphorus is extracellular with the major phosphate store being in bone and the intracellular compartment. The normal range of serum phosphorus concentration in the serum is between 2.2 and 4.4 mg/dL, of which about 55 % is in an ionized form that is physiologically active. The serum phosphate concentration is a poor indicator of the total body phosphorus and rapid shifts of phosphate between the extracellular and intracellular compartments only confound this situation. Interpretation of the serum phosphate is further complicated by a normal diurnal variation, which may be as large as 0.5 mg/dL. However, hypophosphatemia may cause severe life threatening complications, particularly in patients with depleted phosphate stores.
Causes of severe hypophosphatemia
alcohol abuse and withdrawal
refeeding after starvation
respiratory alkalosis
malabsorption
oral phosphate binders
TPN
severe burns
therapy of diabetic ketoacidosis
There is a poor correlation between serum phosphate levels and symptoms. Although hypophosphatemia becomes life threatening when the serum levels are less than 1 mg/dL, symptoms may develop when the serum phosphate is less than 2 mg/dL.
Manifestations include:
myocardial depression
weakness, rhabdomyolysis and respiratory failureRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree





