(1)
Division of Pulmonary and Critical Care Medicine, Eastern Virginia Medical School, Norfolk, VA, USA
Keywords
Liver failureCirrhosisHepatic encephalopathyEsophageal varicesAcute liver failureAcetaminophenAscitesSpontaneous bacterial peritonitis (SBP)Hepatorenal syndromeAlbuminRifaximinLactulosePortal hypertensionBilirubinChronic Liver Failure
Chronic liver failure (CLF) and cirrhosis accounted for more than 26,000 deaths and more than half a million hospitalizations in the United States in 2004, making liver disease the 12th leading cause of death [1]. Hospital and/or ICU mortality rates of cirrhotic patients admitted to the ICU range from 34 to 86 % [2]. The Child-Turcotte-Pugh (CTP) scoring system classifies CLF into three categories based on severity (see Table 34.1). A total CTP score of 5–6 is Child’s class A, well-compensated disease; a CTP score of 7–9 is Child’s class B, in which there is significant functional compromise; and a CTP score of 10–15 is Child’s class C, advanced decompensated disease [3]. The model for end-stage liver disease (MELD) score provides another classification of the severity of chronic liver failure based on the readily obtainable laboratory values of serum creatinine, total bilirubin, and prothrombin time, expressed as the international normalized ratio [3, 4]. Several variations to MELD that include serum sodium (MELD-Na) and the Integrated Model for End-stage liver disease (iMELD) have been shown to improve mortality prediction in cirrhotic patients awaiting liver transplantation [5, 6].
Table 34.1
The Child-Turcotte-Pugh (CTP) scoring system
Parameter | 1 | 2 | 3 |
|---|---|---|---|
Ascites | Absent | Easily controlled | Poorly controlled |
Bilirubin (mg/dL) | <2 | 2–3 | >3 |
Albumin (g/dL) | >3.5 | 2.8–3.5 | <2.8 |
INR | <1.7 | 1.7–2.3 | >2.3 |
Encephalopathy | None | Grade 1–2 | Grade 3–4 |
Cirrhosis is defined histologically as an advanced form of progressive hepatic fibrosis with distortion of the hepatic architecture and regenerative nodule formation. It may be due to a variety of causes. The major clinical consequences of cirrhosis are impaired hepatocyte function, an increased intrahepatic resistance (portal hypertension), and the development of hepatocellular carcinoma. The general circulatory abnormalities in cirrhosis (splanchnic vasodilation, vasoconstriction and hypoperfusion of kidneys, water and salt retention, increased cardiac output) are intimately linked to the hepatic vascular alterations and resulting portal hypertension. The clinical picture of chronic liver disease is frequently dominated by the complications of portal hypertension. In addition, infectious complications are common and associated with worsening of hepatocyte function and portal hypertension.
Causes of Cirrhosis
Viral/infectious
Viral/infectious
Hepatitis B
Hepatitis C
Schistosomiasis
Metabolic/toxic
Alcohol
Toxins
medications
Hereditary hemochromatosis
Wilson’s disease
Nonalcoholic steatohepatitis (NASH)
Autoimmune hepatitis
Cholestatic
Primary biliary cirrhosis
Primary sclerosing cholangitis
Secondary biliary cirrhosis
Vascular
Right heart failure
Budd–Chiari syndrome
Alpha-1-antitrypsin deficiency
Sarcoidosis
Cystic fibrosis
Cirrhosis represents a clinical spectrum, ranging from asymptomatic liver disease to hepatic decompensation. Manifestations of hepatic decompensation include
variceal bleeding
ascites with spontaneous bacterial peritonitis
hepatic encephalopathy
hepatorenal syndrome
hepatopulmonary syndrome
portopulmonary hypertension
hepatocellular carcinoma
hepatoadrenal syndrome
The liver never fails in isolation…it takes each and every organ system down with it!
Metabolic/Hematologic Derangements in Cirrhosis
Hyperglycemia (portal to systemic shunting)
Hypoglycemia (hepatocyte failure)
Hypoalbuminemia
Decreased synthesis of clotting factors prolonged INR
Decreased production of AT, Protein S and C. thrombotic risk
Increased ammonia
Cholestasis
Impaired absorption of fat and fat soluble vitamins
Anemia
Microcytic from iron deficiency
Macrocytic folate and B12 deficiency
Hyponatremia (Na < 135 mmol/l)
An independent predictor of mortality [7]
From increased ADH due to decreased effective circulating volume
Thrombocytopenia
Hypersplenism
Alcohol
Marrow suppression
“Low level” DIC
Renal dysfunction
Impaired immunity
Spontaneous Bacterial Peritonitis
Spontaneous bacterial peritonitis (SBP) is seen in up to 30 % of patients with ascites [8]. Patients who have SBP present with fever, diffuse abdominal pain or tenderness, altered mental status, leukocytosis, or worsening renal function. Approximately 15 % of patients do not have any signs or symptoms of SBP. SBP is believed to be a consequence of increased bacterial translocation largely via the lymphatic system. Small intestinal bacterial overgrowth is frequently present in the advanced stages of liver cirrhosis. Furthermore, cirrhosis is associated with structural and functional alterations in the intestinal mucosa that increase permeability to bacteria and bacterial products [8]. Only a few intestinal bacteria are able to translocate to the mesenteric lymph nodes including Escherichia coli, Klebsiella pneumonia and other Enterobacteriaceae [8]. These are the species most commonly implicated in SBP. The diagnostic test of choice is abdominal paracentesis. An ascitic fluid neutrophil count higher than 250/mm3, in the absence of an intra-abdominal surgical source of infection, such as a perforated peptic ulcer or abscess, is diagnostic of SBP. Fluid cultures are positive in approximately half of the cases. An elevated serum procalcitonin (PCT) level is reported to have a high diagnostic accuracy for SBP, and appears to be a useful adjunctive diagnostic aid [9]. Almost all cases of SBP are secondary to a single micro-organism, with Escherichia coli and Klebsiella pneumoniae accounting for approximately half of the cases [10]. Treatment consists of a 5-day course of a third-generation cephalosporin, such as intravenous ceftriaxone, which usually results in an excellent clinical response with resolution of SBP. For patients unable to take a cephalosporin, intravenous ciprofloxacin, followed by oral administration, is recommended [10].
SBP is associated with the development of hepatorenal syndrome in about 30 % of patients. In 1999, Sort and colleagues reported the results of a RCT in which patients with SBP were randomized to cefotaxime + albumin or cefotaxime alone [11]. 20 % albumin was given at a dose of 1.5 mg/kg at the time of SBP diagnosis and 1.0 mg/kg after 48 h. The study does not report the duration of the albumin infusion. Renal impairment developed in 21 patients in the cefotaxime group (33 %) and 6 in the cefotaxime + albumin group (10 %) (P = 0.002). Eighteen patients (29 %) in the cefotaxime group died in the hospital, as compared with 6 (10 %) in the cefotaxime-plus-albumin group (P = 0.01); at three months, the mortality rates were 41 % (a total of 26 deaths) and 22 % (a total of 14 deaths), respectively (P = 0.03). Salerno and colleagues performed a metaanalysis of studies comparing albumin to placebo in patients with SBP [12]. The meta-analysis included three studies; the incidence of renal impairment in control groups was 44 of 144 (30.6 %), compared with 12 of 144 (8.3 %) in groups given albumin. The pooled odds ratio for a reduction in renal impairment after albumin infusion was 0.21 (95 % CI, 0.11–0.42). Mortality among controls was 51 of 144 (35.4 %), compared with 23 of 144 (16.0 %) among patients who received albumin. The pooled odds ratio for decreased mortality after infusion of albumin was 0.34 (95 % CI, 0.19–0.60). Considering the pharmacodynamics of albumin (reviewed in detail in Chap. 9) we would recommend a “bolus” of 5 % albumin (500–1,000 mL) at the time of diagnosis (given over 1–2 h), followed by a continuous infusion of 20–25 % albumin at a rate of 10–15 mL/h. Alternatively, an infusion of 100 mL of 20–25 % albumin may be given over 3–4 h, followed a continuous infusion at a rate of 10–15 mL/h.
Patients who have had an episode of SBP are at an increased risk of recurrent episodes, hence antibiotic prophylaxis is recommended in these patients. Following an initial episode of SBP, 1-year recurrence rate is 55 % with a 1-year survival is less than 50 %. Norfloxacin has been demonstrated to reduce the recurrence of SBP and is currently regarded as the agent of choice for secondary prophylaxis [13]. Amoxicillin-clavulanate and trimethoprim-sulfamethoxazole are acceptable alternatives in those unable to tolerate quinolones. Primary prophylaxis, defined as antibiotic treatment of patients without prior SBP, has been suggested in patients who have chronic liver failure and ascites who fulfill the following criteria:
ascitic fluid protein concentration lower than 1 g/dL
serum bilirubin level higher than 3.2 mg/dL, and
platelet count higher than 98,000/mm3, as these patients have a threefold-increased risk of developing SBP within 1 year.
A RCT demonstrated that primary prophylaxis with norfloxacin significantly reduced the 1-year risk of developing SBP when compared with placebo (7 % versus 61 %; P < 0.001) [14]. A meta-analysis which included both primary and secondary prophylaxis demonstrated a lower incidence of infections (RR 0.32; 95 % CI 0.20–0.51) with an overall mortality benefit (RR 0.65; 95 % CI 0.48–0.88) [15].
Acid-suppressive therapy (PPI’s and H2RB’s) should be avoided in patients with cirrhosis as acid-suppressive therapy has been demonstrated the increase the risk of SBP [16]. Acid suppressive therapy is associated with increased colonization of the small bowel. Presumably this leads to increased bacterial translocation with an increased risk of SBP.
Hepatic Encephalopathy
Hepatic encephalopathy (HE) is a significant neuropsychiatric syndrome that most commonly occurs in decompensated cirrhosis [17]. Clinical features range from clinically imperceptible symptoms in minimal HE, which require neuropsychometric testing to identify, to a comatose state in the worst cases. Many factors have been implicated in its pathogenesis, including derangements in neurotransmitter pathways, cerebral blood flow modulation, and systemic inflammatory responses. The ammonia hypothesis states that impaired hepatic breakdown of ammonia results in multiple neurotoxic effects, including altering the transit of amino acids, water, and electrolytes across the neuronal membrane and propagating astrocyte swelling and cerebral edema. Contrary to “classic teaching”, Ong and colleagues found a good correlation between the serum ammonia levels and the severity of hepatic encephalopathy [18]. Furthermore, there was no significant difference between venous and arterial ammonia levels. The Working Party for Hepatic Encephalopathy established nomenclature for hepatic encephalopathy in 1998 [19]. Type A HE refers to HE secondary to acute liver failure, type B refers to enteric hyperammonemia (without liver disease), and type C is associated with chronic liver disease. The severity of HE is graded using the West Haven criteria (grades 1–4) [17].
Grades of Hepatic Encephalopathy
Grade 0
◦ No signs or symptoms
Grade 1
◦ Trivial lack of awareness, euphoria or anxiety, shortened attention span impaired performance of addition
Grade 2
◦ Lethargy or apathy, minimal disorientation for time or place, subtle personality change, inappropriate behavior, impaired performance of subtraction
Grade 3
◦ Somnolence to semi-stupor, but responsive to verbal stimuli, confusion, gross disorientation
Grade 4 Coma (unresponsive to verbal or noxious stimuli)
Hepatic encephalopathy develops in up to 50 % of patients with cirrhosis and is a feature of decompensated cirrhosis. Hepatic encephalopathy portends a worse survival for patients compared with similar patients without HE. Acute worsening of HE should prompt an evaluation for reversible causes, such as gastrointestinal bleeding, hypovolemia, hypoglycemia, hypokalemic metabolic alkalosis, infection, constipation, hypoxia, or excessive use of sedatives
Colonic bacteria with urease activity produce ammonia in the gut. The disaccharide lactulose (β-galactosidofructose) is the mainstay of induction and maintenance treatment of HE [17]. The microvilli of the small bowel lack disaccharides capable of breaking down lactulose, permitting the entry of this disaccharide into the colon. Lactulose is broken down by bacterial flora to short chain fatty acids lowering colonic pH converting NH3 to nonabsorbable NH4+ which remains in the colon, reducing plasma ammonia. In addition, lactulose shifts the colonic flora from urease to non-urease producing bacterial species and reduces bacterial load by its cathartic effect. The starting dose of lactulose is commonly 30 g twice a day, titrated to two to three soft stools per day. A meta-analysis performed in 2004 found that nonabsorbable disaccharides were superior to placebo but did not improve survival [20]. When only high-quality trials were included in this meta-analysis, nonabsorbable disaccharides had no effect on HE. Despite the lack of high level evidence clinical guidelines recommend lactulose as first-line therapy [21].
Nonabsorbable antibiotics such as rifaximin are frequently added to lactulose to further decrease intestinal ammonia production [22]. Rifaximin is however not FDA approved for the treatment of episodic HE, only for the secondary prevention of HE. Sharma et al. conducted a RCT (n = 120) comparing rifaximin and lactulose with lactulose and placebo in patients with HE [23]. Patients in the lactulose and rifaximin group had a higher proportion of complete reversal of HE (76 % vs 50.8 %, P < 0.004), shorter hospital stay, and a striking improvement in 10-day mortality (49.1 % vs 23.8 %, P < 0.05). Probiotics and prebiotics (Bifidobacterium and fructose-oligosaccharides) which alter the bowel flora favoring non-urease producing bacterial species have been demonstrated to lower ammonia levels and improve mentation in patients with hepatic encephalopathy [24–26]. Most patients require maintenance medications (lactulose and/or rifaximin) at the time of hospital discharge as secondary prophylaxis for HE [27, 28].
Previously, aggressive protein restriction was recommended; however, this is now believed to worsen the nutritional status of patients and decreases overall survival [29]. Guidelines published by the European Society for Clinical Nutrition and Metabolism (ESPEN) recommended that patients with cirrhosis should have an energy intake of 35–40 kcal/kg body weight per day and a protein intake of 1.2–1.5 g/kg body weight per day [30]. Zinc deficiency impairs the activity of urea cycle enzymes and glutamine synthetase. Zinc deficiency has been implicated in the pathogenesis of hepatic encephalopathy as diminished serum zinc levels and their inverse correlation with blood ammonia levels have been reported [31]. Zinc supplementation in the treatment of hepatic encephalopathy is based on a small number of controlled studies that provided inconsistent results regarding efficacy, types and doses of zinc used, and duration of therapy [32].
Molecular Adsorbent Recirculating System (MARS) was introduced in 1999 and is based on the concept of albumin dialysis. This system was designed to remove protein- and albumin bound toxins, however it also removes non-protein bound molecules such as ammonia [17]. While MARS has been demonstrated to improve HE it has not been demonstrated to improve survival [33].
Hepatorenal Syndrome
Hepatorenal syndrome (HRS) is a functional form of renal failure that occurs in patients with end-stage liver disease. The pathophysiological hallmark of HRS is vasoconstriction of the renal circulation. The mechanism of the vasoconstriction is incompletely understood; it may be multifactorial, involving disturbances in the circulatory function and activity of the systemic and renal vasoactive mechanisms. Patients with HRS demonstrate arterial vasodilation in the splanchnic circulation with arterial constriction in the cerebral, hepatic and renal vascular beds. Furthermore, patients’ who develop HRS appear to have inadequate cardiac output. Non-azotemic cirrhotic patients who develop HRS have lower cardiac output and higher plasma renin activity (PRA) than those who don’t progress to HRS [34]. These findings support the hypothesis that a hyperdynamic circulation is essential to maintain renal perfusion. When cardiac output deceases effective hypovolemia occurs, leading to renal hypoperfusion and HRS [35]. In 1996 the International Ascites Club established diagnostic criteria for the diagnosis of HRS [36]. HRS was further classified as type 1 and type 2 according to the rate of decline of renal function. Type 1 was arbitrarily defined as a 100 % increase in serum creatinine reaching a value of greater than 1.5 mg/dL in less than 2 weeks. Patients who had a slower decline in renal function were deemed to have type 2 HRS. The current diagnostic criteria for HRS (proposed originally by the International Ascites Club in 1996 and revised in 2005) are listed below [35, 36].
Hepatorenal Syndrome: Diagnostic Criteria
Presence of cirrhosis with ascites
Serum creatinine >1.5 mg/dL
Failure of serum creatinine to improve to <1.5 mg/dL after cessation of ALL diuretics + volume expansion with intravenous albumin (see section on SBP)
HRS can be diagnosed in the presence of infection if shock is absent
NO current or recent use of nephrotoxic drugs
No evidence of parenchymal kidney disease; proteinuria <500 mg/d; <50 RBC high power field
Diagnostic Approach
Urinalysis, including urine Na and fractional Na excretion
Rule out abdominal compartment syndrome (paracentesis and albumin infusion)
Rule out SBP (paracentesis and albumin infusion)
Renal ultrasound to exclude hydronephrosis/obstructive uropathy
In a prospective cohort study of non-azotemic cirrhotic patients with ascites the incidence of HRS was reported as 18 % at 1 year and 39 % at 5 years [37]. Patients with type 1 HRS have a very poor prognosis compared to patients with type 2 HRS. The median survival time for type 1 HRS has been reported to be 14 days. Common precipitating factors for type 1 HRS include bacterial infections, particularly SBP, variceal hemorrhage, major surgery and alcoholic hepatitis. Excessive diuresis and the use of contrast agents have also been implicated in precipitating HRS. Tsai et al. demonstrated that 80 % of patients with HRS precipitated by sepsis had adrenal dysfunction (see hepato-adrenal syndrome) [38]. Replacement therapy with hydrocortisone improves systemic hemodynamics and in-hospital survival [39]. The only effective medical therapy currently available for the management of HRS is the administration of vasoconstrictors together with volume expansion with albumin. Volume expansion with albumin and vasopressin analogues (ornipressin and terlipressin), norepinephrine, midodrine and somatostatin (octreotide) have been used with variable success [40, 41]. Liver transplantation is considered the treatment of choice for patients with cirrhosis and type 1 HRS because it “allows for both the liver disease and associated renal failure to be cured.”
HRS is a frequent complication of over overdiuresis (with Lasix), particularly in the setting of a contrast study or a therapeutic paracentesis. Patients who undergo a contrast study and or paracentesis require fluid loading (with albumin).
LASIX is NOT a volume expander; its use may lead to a fatal outcome in patients with cirrhosis.
Treatment of HRS
Renal vasodilators (low dose dopamine, intravenous prostaglandin-E, endothelin antagonists) have no proven role in the treatment of HRS. Reversing splanchnic and peripheral vasodilation with a vasoconstrictor combined with an albumin infusion is currently the cornerstone of treatment of HRS. This strategy is based on a number of small case series and clinical trials [42–46]. Reversal of HRS has been reported in 25–83 % of patients treated with pressor agents plus albumin compared to 8.7–12.5 % of patients treated with albumin alone [47]. However, long term patient survival without liver transplantation is not improved. Albumin alone does not appear to reverse HRS. Furthermore, normal saline should not be used as a volume expander in cirrhotic patients with a decline in renal function. Terlipressin is the most studies vasopressor agent for the treatment of HRS. This synthetic vasopressin derivative is believed to have a much greater effect on vascular receptors (V1) than renal vasopressin receptors (V2). A metaanalysis of ten clinical trials of terlipressin reported that this therapy was well tolerated and reversed HRS in 52 % of cases [48]. Norepinephrine plus albumin appears to be a useful alternative to terlipressin [49].
Combination Therapy for HRS
Intravenous terlipressin (not yet approved for use in the US) initial dose of 0.5–1 mg every 4–6 h, increased on day 4 to 2 mg 4–6 hourly if serum creatinine has not decreased by >30 % from baseline + intravenous albumin (see section on SBP)
Intravenous norepinephrine; start at a dose of 0.01 μg/kg/min and titrate to increase the MAP by 10–15 mmHg (even in the absence of hypotension) + intravenous albumin
Oral midodrine initial dose of 7.5 mg TID titrate up to 15 mg TID to increase MAP by 10–15 mmHg + octreotide 100 μg SQ TID increased to 200 μg TID on day 2 if renal function has not improved + intravenous albumin. The use of octreotide alone without midodrine has not been shown to be effective.
Hepato-adrenal Syndrome
Sepsis and end-stage liver disease have a number of pathophysiologic mechanisms in common (endotoxemia, increased levels of pro-inflammatory mediators, decreased levels of HDL), and it is therefore not surprising that adrenal insufficiency is common in patients with end-stage liver disease [50]. Furthermore, sepsis in the cirrhotic patient likely increases the risk of adrenal insufficiency. Tsai and colleagues performed a corticotrophin stimulation test in 101 patients with cirrhosis and sepsis [38]. In this study 51.4 % of the patients were diagnosed with adrenal insufficiency; survival at 90 days was 15.3 % in these patients compared to 63.2 % in those patients with normal adrenal function. Fernandez and co-authors compared the survival of patients with cirrhosis and sepsis who underwent adrenal function testing in which patients with adrenal insufficiency were treated with hydrocortisone (Group 1) compared to a control group (Group 2) that did not undergo a cosyntropin testing and were not treated with corticosteroids [39]. The incidence of adrenal failure was 68 % in Group 1; the hospital survival was 64 % in Group 1 as compared to 32 % in Group 2 (p = 0.003). These data suggest that adrenal dysfunction is common in critically ill patients with end-stage liver disease and that treatment with corticosteroids may improve outcome.
Pulmonary Consequences of Portal Hypertension
Two distinct pulmonary vascular disorders can occur in cirrhosis: hepatopulmonary syndrome (HPS) and portopulmonary hypertension (POPH). Both can coexist in the same patient. Portopulmonary hypertension is seen in 0.5–5 % of patients with cirrhosis and/or portal hypertension and presents in similar ways to patients with pulmonary hypertension from other causes. Diagnosis is established by echocardiography and right heart catheterization. Recent case reports and series have demonstrated improvement of pulmonary hypertension with oral sildenafil [51]. HPS is defined as a defect in arterial oxygenation induced by intrapulmonary vascular dilatations. This entity is seen in 8–17 % of patients with cirrhosis, and median survival is 11 months. Liver transplantation resolves HPS in the majority of patients
Infection and Cirrhosis
Cirrhotic patients have several abnormalities which increase the susceptibility to bacterial infection; these include deficiency of bactericidal and opsonic activities, impaired monocyte function, depressed phagocytic activity, defective chemotaxis, and low levels of serum complement. A National Hospital Discharge Survey demonstrated that hospitalized cirrhotics are more likely to develop sepsis (RR 2.6) and to die from sepsis (RR 2.0) than hospitalized non-cirrhotics [52]. Fifteen to 35 % of cirrhotics develop nosocomial infection, with infection accounting for 30–50 % of deaths in patients with cirrhosis.
Usual infections
◦ SBP
◦ Pneumonia
◦ Gram-negative bacteremia
◦ UTI
Common pathogens
◦ Escherichia coli
◦ Staphylococcus aureus
◦ Enterococcus faecalis
◦ Streptococcus pneumoniae
◦ Pseudomonas aeruginosa
Diagnosis of infection is difficult in patients with cirrhosis
◦ reduced WBC due to hypersplenism
◦ tachycardia and hyperdynamic circulation
◦ hyperventilation due to hepatic encephalopathy
◦ blunted febrile response
◦ Cirrhosis itself results in low grade fever
In patients with cirrhosis, infections
Further impair systemic and splanchnic hemodynamics
Impair coagulation
Worsen liver function
May trigger variceal bleed
Supportive Care of the Hospitalized Cirrhotic
Do not replace clotting factors or platelets unless bleeding
Feed with a 1 g/kg protein diet
SCD’s for DVT prophylaxis? heparin 500 sc q 12 or LMWH
Exclude infection; culture blood, ascitic fluid
Prophylactic antibiotics in high risk patients
DO NOT DIURESE… give 25 % albumin
Paracentesis in patients with tense ascites (replace albumin)
Avoid nephrotoxic drugs
AVOID sedatives, particularly benzodiazepines which can precipitate hepatic encephalopathy. Haloperidol and dexmedetomidine are okay.
Lactulose for regular stool
Monitor venous ammonia
ACTH stim test replace with hydrocortisone if adrenal insufficiency
Ultrasound + Doppler to exclude portal/splenic vein thrombosis and hepatocellular carcinoma
α feto-protein level
The Coagulopathy of Chronic Liver Disease
Classic teaching suggests that the coagulopathy of liver disease leads to an increased risk of bleeding. However recent data suggests that this dogma is likely incorrect and that chronic liver disease results in a procoagulant state. The concept that patients with liver disease are “auto-anticoagulated” should be abandoned. Patients with cirrhosis have multiple defects in the coagulation pathway including deficiencies in both procoagulant and anticoagulant factors, thrombocytopenia, hypofibrinogenemia and hyperfibrinolysis [53]. These derangements reflected in conventional coagulation indices (low platelet count, increased INR, low fibrinogen) have led cirrhosis to become regarded as a hemorrhagic disorder. Cirrhosis is characterized by a reduction in synthesis of all the procoagulant factors, which worsens as the disease progresses [53]. The exception to this is the powerful procoagulant factor VIII, which is synthesized in extra-hepatic sites (vascular endothelium) and shows the opposite trend. Furthermore, the synthesis of the anti-coagulants Protein S and Protein C is reduced (promoting a pro-thrombotic state). In addition, elevated levels of antiphospholipid antibodies are detected in some patients with cirrhosis [54]. Thrombocytopenia is common owing to a reduction in platelet production sequestration in the spleen and increased turnover. Although platelet numbers are low this may be compensated by platelet hyper-activation owing to increased levels of platelet protein adhesion factor and von Willebrand factor and reduced levels of ADAMTS13 (von Willebrand factor cleaving protease) [53]. A reevaluation of the disturbed coagulation pathways in cirrhosis has now led cirrhosis to be considered a pro-coagulant state. More accurate tests of coagulation including thrombin generation assays have shown that the plasma from patients with cirrhosis appears to be hypercoagulable in comparison with healthy controls [55, 56].
The factors promoting bleeding and/or thrombosis in patients with cirrhosis are listed in Table 34.2. The reader is referred to an excellent review on this topic by Tripodi and Mannucci [57]. Chronic liver disease results in a balanced reduction of both the procoagulant and anticoagulant pathways. However, although rebalanced, the coagulation system in patients with chronic liver disease is not as stable as that in healthy persons, who have an excess of both procoagulants and anticoagulants. Therefore, the relative deficiency of both coagulation-system drivers makes the balance fragile in patients with liver disease and may tip it toward hemorrhage or thrombosis, depending on the prevailing circumstantial risk factors [57]. The disturbed coagulation system in patients with cirrhosis may therefore lead to bleeding, thrombosis or even both depending on the trigger (sepsis, renal dysfunction, hypovolemia). Although still common practice, the use of FFP to correct the “coagulopathy” of liver disease should be abandoned, even in patients undergoing invasive procedures. The American Association for the study of Liver Diseases (AASLD) warns against the indiscriminant use of FFP prior to paracentesis, central line placement and liver biopsies (see Chap. 7) [58, 59].
Favors bleeding | Favors thrombosis |
|---|---|
Decreased procoagulant factors | Deceased anticoagulant factors |
Decreased platelet number | Increased von-Willebrand factor |
Decreased platelet function | Decreased ADAMTS 13 |
Decreased fibrinogen | Increased factor VIII |
Increased fibrinolysis | Increased procoagulant microvesicles |
Decreased red call mass | Lupus anticoagulant |
Recent studies have documented an increased risk of venous thrombosis in patients with cirrhosis. Dabbagh et al. reported that 6.3 % of hospitalized patients with chronic liver disease developed venous thromboembolism [60]. In this study the risk of VTE was similar between INR quartiles, with half the cases occurring in patients with an INR > 1.6. VTE prophylaxis was not used in 75 % of patients. In a case-matched study of patients admitted to a tertiary care center Gulley et al. reported a higher incidence of VTE in cirrhotic patients as compared to controls (1.8 % vs. 0.9 %, P = 0.007) [61]. Using data from the Nationwide Inpatient Sample in 2005, Ali et al. reported that 1.8 % of hospitalizations in cirrhotic patients were for VTE [62]. The optimal strategy to prevent DVT in hospitalized patients with cirrhosis is unclear. This subject was not addressed in the most recent ninth edition of the American College of Chest Physicians Evidence-Based Clinical Practice Guidelines [63]. Since these are high risk patients with a thrombophilia, SCD’s together with pharmacologic prophylaxis (heparin or LMWH) may be preferred in non-bleeding patients while SCD’s should be used in bleeding patients (esophageal varices). LMWH appears to be safe in patients with cirrhosis and hepatic dysfunction (see below). Patients with cirrhosis are at an increased risk of portal vein thrombosis. This is reviewed below.
Thrombin, the main effector of the coagulation cascade, elicits a number of cellular effects through a family of G-protein coupled receptors known as protease activated receptors [53]. Excessive thrombin generation with activation of these receptors has been implicated in the increased fibrogenesis seen in patients with cirrhosis. Anticoagulants, which decrease thrombin may therefore reduce fibrogenesis in patients with cirrhosis (see below).
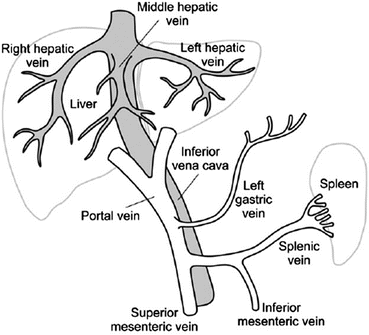
Portal Vein Thrombosis
The portal vein originating from the confluence of the superior mesenteric and splenic veins, accounts for 75 % of the blood supply to the liver (see Fig. 34.1) [64]. Portal vein thrombosis (PVT) is an important complication in patients with chronic liver disease. The incidence of PVT varies between 0.6 and 16 % in patients with well-compensated cirrhosis to 35 % in patients with advanced liver disease particularly those with hepatocellular cancer [64, 65]. PVT in the absence of cirrhosis accounts for about 5–10 % of cases of portal hypertension in the Western hemisphere; most of these cases are related to an inherited or acquired thrombophilia. In developing countries PVT is a common cause of portal hypertension. In candidates for liver transplantation, the patency of the portal vein is crucial. Partial PVT is likely to make anastomoses technically difficult and increases the risk of post-transplant complications including complete PVT. Complete PVT is a contraindication for transplantation [65].