Fig. 17.1
The influence of epidural ropivacaine dose (a) and the number of blocked epidural segments (b) on the elimination clearance (CL) of propofol. An epidural dose of 150 mg ropivacaine decreases the propofol clearance from 2.58 to 2.0 L/min; 20 blocked epidural segments reduce the clearance of propofol from 2.64 to 1.87 L/min. The discontinuous line shows the 95 % confidence intervals. The dots are the empirical Bayesian estimates of clearance for each patient (Adapted from Sitsen et al. [42] With permission from Wolters Kluwer Health, Inc.)
Pharmacokinetic Interactions Between Anesthetics and Concomitant Home Medication
Apart from the pharmacokinetic interactions between the agents given perioperatively, anesthetic agent pharmacology may be influenced by agents that patients use preoperatively at home. Phase I drug elimination includes oxidation, hydrolysis, and reduction of the parent drug. Oxidation is generally catalyzed by one or more of the nearly 60 cytochrome P450 members of this isoenzyme family. For the metabolic clearance of drugs, cytochrome P450 3A4 is probably the most important cytochrome isoenzyme. This subenzyme is, for example, important for the oxidation of midazolam and alfentanil . Other important subenzymes for anesthetic agent breakdown are CYP 2B6, 3A3, 3A5, and 2E1. The 60 isoenzymes are subject to induction and inhibition, causing a possible increase or decrease of their catalyzing capacity, thus affecting the clearance of agents and thereby drug effect. Many agents are known to be potent cytochrome P450 inducers and cytochrome P450 inhibitors and may greatly alter the dose–concentration time relationship of sedatives and opioid analgesics. The clinical impact of enzyme induction and inhibition may be huge. Whereas anesthetic–anesthetic drug interactions generally cause plasma concentration changes in the order of 25 %, certain antibiotics, antifungal agents, or anticonvulsants may increase or decrease anesthetic opioid or sedative concentrations by two- to tenfold through enzyme induction or inhibition.
The clinical consequences of cytochrome P450 inhibition and induction thus may be significant. Numerous case reports describe the unpleasant effects of enzyme inhibitors and/or inducers on opioid effect. Discontinuation of carbamazepine and thus termination of the cytochrome enzyme-inducing effect caused a woman taking methadone, 210 mg PO daily for lung cancer pain relief, to lose consciousness and experience severe respiratory depression 11 days later [44]. Similarly, a male and female patient taking methadone per os on a daily basis went into coma and respiratory depression several days after the start of the intravenous administration of fluconazole and cimetidine, respectively, both being cytochrome P450 3A4 inhibitors [45, 46].
Cytochrome P450 Enzyme Inhibition
Inhibition of cytochrome P450 by one agent may lead to a reduced clearance and thereby to an increase in desired effects and adverse effects of a second agent. The increase in adverse effects may lead to potentially hazardous consequences. Enzyme inhibition occurs when a cytochrome enzyme is unable to metabolize its substrate due to the interference of another substrate [47]. Often this is the case when the cytochrome simply reaches its maximal metabolizing potential, is competitively blocked, or results from a negative feedback induced by a substrate [47]. Enzyme inhibition develops fast, sometimes within 24 h, and may last for weeks after the discontinuation, for example, as in the case of cimetidine [48, 49]. Well-known cytochrome P450 enzyme inhibitors include macrolide antibiotics like erythromycin and troleandomycin , antifungal agents like ketoconazole, Ca-channel blockers like verapamil and diltiazem, anticancer agents like tamoxifen, and omeprazole, cimetidine, grapefruit juice, and propofol [50]. Except for remifentanil, the clearance of opioids is, besides redistribution, predominantly dependent on hepatic metabolism. Drug interactions with opioids involving cytochrome inhibition may cause significant drug concentration changes and may therefore be of great clinical relevance.
Erythromycin and troleandomycin cause significant inhibition of CYP 3A4 through the formation of an inactive complex. The result is that with increasing erythromycin intake over days, the clearance of alfentanil gradually decreases, causing the plasma alfentanil concentration, after a similar dose, to increase. The clearance of alfentanil may decrease by 25 % with an increase in the elimination half-life of over 50 % [51]. Alfentanil thus shows prolonged activity in the presence of erythromycin, causing prolonged analgesia but also prolonged respiratory depression [51, 52]. Similarly, the available drug concentration of midazolam over time after oral midazolam intake increases by a factor of 3.8 after pretreatment with oral erythromycin, 500 mg for 5 days [53, 54]. Olkkola and colleagues reported the potential harmful interaction between erythromycin and midazolam , observing prolonged sedation and amnesia after the intake of erythromycin 3 times daily 500 mg for a week [55]. Similarly, an 8-year-old boy lost consciousness during an erythromycin infusion after premedication with midazolam as a result of unexpectedly high plasma midazolam concentrations due to cytochrome P450 inhibition [56].
Antifungal agents are frequently used in the intensive care for the treatment and prevention of fungal infections. Antimycotics like fluconazole, ketoconazole, and itraconazole all strongly inhibit cytochrome P450 3A [57]. Ketoconazole is the most potent inhibitor, predominantly on CYP 3A. CYP 3A is the most important cytochrome for catalyzation of drug metabolism and accounts for 40–50 % of cytochrome activity in the liver but is also present in the small intestine. Antifungal agent use in ICU patients may strongly prolong the effect of midazolam with plasma peak midazolam concentrations increasing by three- to fourfold [58]. Sedation with midazolam in the presence of azole agents in ICU patients thus may cause hazardous consequences. Also ketamine that is metabolized through CYP 2B6, 3A4, and 2C9 may be subject to a clearance reduction by the concomitant use of antifungal agents. The same is true for alfentanil , fentanyl, and oxycodone. No data suggest that propofol, which is subject to metabolism through CYP 2B6 and 2C, interacts with antifungal agents . The calcium-channel blockers verapamil and diltiazem show similar interaction to the antifungal agents and also affect the pharmacokinetics of benzodiazepines [59].
Lastly, grapefruit juice is also known as a potent cytochrome P450 inhibitor. Several studies report on the inhibition of benzodiazepine clearance through grapefruit juice [60–62]. Grapefruit juice has been reported to increase the plasma concentration of oral midazolam by over 50 %. Apart from grape fruit juice, many other fruits inhibit cytochrome activity. The inhibitory potential of human cytochrome P450 3A is in the order: grapefruit > black mulberry > wild grape > pomegranate > black raspberry [63]. All the abovementioned fruit juices are thus capable of inhibiting midazolam 1-hydroxylation. These fruit juices may thus potentiate the effect of oral benzodiazepines when used for sleep medication in the home situation or as premedication for surgery. Grapefruit juice also increases the area under the curve of S-ketamine by threefold [64].
Other inhibitors of cytochrome enzymes include the proton pump inhibitors like omeprazole and pantoprazole. Diazepam and midazolam are substrates for CYP 2C19, which is inhibited by omeprazole [65]. In a similar manner, the H2-receptor antagonist cimetidine affects the plasma midazolam concentrations. Cimetidine co-medication results in a significant increase in the plasma midazolam concentration by about 30 % [66]. The most potent now known inhibitor is ritonavir, a protease inhibitor used as an antiretroviral agent for the treatment of HIV infection. Ritonavir leads to an enormous increase in midazolam and alfentanil effect with an increase in the area under the curve of both agents in the order of 25- to 30-fold [67]. Anesthesiologists should be aware of this interaction when opioids or benzodiazepines are used in the presence of ritonavir.
Cytochrome P450 Enzyme Induction
Enzyme induction is defined as the process of an increase in metabolizing enzyme quantity or activity as the result of the administration of another agent. Induction of a cytochrome P450 subenzyme by one agent may lead to an increased clearance and thereby to a reduced effect of a second agent (Fig. 17.2), or it may lead to an unexpected inactivation or detoxification. The sometimes unexpected decrease in effect may lead to potentially hazardous consequences when the agent is not titrated to effect. Enzyme induction is dose and agent dependent, often occurring at the higher ranges of clinical drug administration. Enzyme induction involves receptor binding, gene encoding, mRNA transcription, and synthesis of the new protein and may be effective within hours after administration [47, 68]. Well-known cytochrome P450 enzyme inducers include (Table 17.1) barbiturates, tobacco smoke, antiepileptics like phenytoin and carbamazepine , rifampicin, and ethanol [50]. Enzyme induction may also be caused by ingestion of char-grilled meat, dexamethasone, St John’s wort, alcohol, or by obesity and fasting [47]. The influence of enzyme induction may be significant. The enhanced clearance may lead to a tenfold reduction in the plasma concentration of a drug leading to close to no effect at a normal drug dose and an area under the curve of only 0.3–0.4 of the control. In Fig. 17.2 illustrates the influence of the cytochrome P450 inducer rifampicin on the concentration–time relationships of midazolam and triazolam after oral intake of clinically significant doses of 15 mg and 0.5 mg, respectively. In the absence of rifampicin, these benzodiazepine dosages assure hypnosis or deep sedation, while with rifampicin, the plasma concentration of these agents drop dramatically, leaving the patients awake [69].
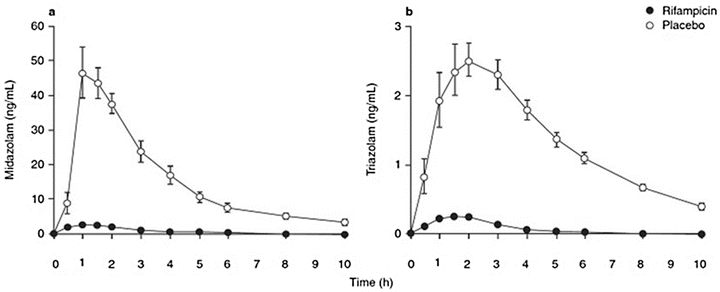
Fig. 17.2
Mean plasma midazolam (a) and triazolam concentration (b) after a 15 mg oral midazolam dose or an 0.5 mg oral triazolam dose, after treatment with placebo or rifampicin 600 mg daily, for 5 days (Adapted from Niemi et al. [69] with permission from Springer; Backman et al. [91] with permission from John Wiley and Sons; and Villikka et al. [92] with permission from John Wiley and Sons)
Table 17.1
Enzymes, substrates, inhibitors, and inducers of cytochrome P450 enzymes relevant to anesthetic practice
CYP subenzyme | Substrate | Inhibitors | Inducers |
|---|---|---|---|
CYP 1A2 | Caffeine | Cimetidine | Smoking |
Haloperidol | Erythromycin | Phenytoin | |
Theophylline | Grapefruit juice | Phenobarbitone | |
Paracetamol | Amiodarone | Omeprazole | |
Lignocaine | Insulin | ||
Ondansetron | Char-grilled meat | ||
Ropivacaine | Ketamine | ||
Naproxen | |||
Amitriptyline | |||
CYP 2C9 | Phenytoin | Ketoconazole | Rifampicin |
Irbesartan | Fluconazole | Phenobarbitone | |
Losartan | Metronidazole | ||
Amitriptyline | Amiodarone | ||
Ibuprofen | Lovastatin | ||
Diclofenac | |||
CYP 2C19 | Diazepam | Cimetidine | Rifampicin |
Phenytoin | Omeprazole | Phenobarbitone | |
Omeprazole | Indomethacin | ||
Pantoprazole | Ketoconazole | ||
Propranolol | |||
Indomethacin | |||
Progesterone | |||
CYP 2D6 | Haloperidol | Cimetidine | Dexamethasone |
Amitriptyline | Ranitidine | Rifampicin | |
Flecainide | Chlorpromazine | Tramadol | |
Codeine | Amiodarone | ||
Lignocaine | Celecoxib | ||
Metoclopramide | Methadone | ||
Ondansetron | |||
Tramadol | |||
CYP 2E1 | Alcohol | Disulfiram | Acetone |
Enflurane | Alcohol | ||
Halothane | Isoniazid | ||
Isoflurane | Obesity | ||
Sevoflurane | Fasting | ||
Paracetamol | Ketamine | ||
Ropivacaine | |||
Theophylline | |||
CYP 3A4 | Nifedipine | Grapefruit juice | Rifampicin |
Diltiazem | Erythromycin | Carbamazepine | |
Amiodarone | Clarithromycin | Phenytoin | |
Fentanyl | Ciprofloxacin | Phenobarbitone | |
Alfentanil | Fluconazole | St John’s wort | |
Sufentanil | Ketoconazole | Ketamine | |
Codeine | Cimetidine | ||
Lignocaine | Propofol | ||
Bupivacaine | Verapamil | ||
Ropivacaine | |||
Midazolam | |||
Triazolam | |||
Diazepam |
Hepatic enzyme induction is furthermore important for the metabolism of inhalational agents. Cytochrome P450 2E1 catalyzes the oxidation of halothane to trifluoroacetyl. This compound is excreted through the urine, but some binds to hapten and may result in halothane-induced hepatitis. Hepatic enzyme inducers may induce P450 2E1 and enhance the formation of this hepatotoxic compound during halothane anesthesia, whereas cimetidine, through enzyme inhibition, may protect against halothane-associated hepatotoxicity [70].
Pharmacodynamic Interactions Between Anesthetics
Because of the small therapeutic window, a detailed characterization of the concentration–effect relationships of anesthetic agents and their interactions is required to allow a proper selection of the various intravenous agents and their combinations, to obtain an optimal therapeutic pharmacological effect while avoiding significant side effects [5].
Bovill reviewed the methodology of the analysis of drug interaction and described four ways of interaction analysis: fractional analysis, isobolographic analysis, method of Plummer and Short, and the parallel line assay [71]. From these, isobolographic analysis and, in parallel, response surface modeling have become the most important methods of analysis of pharmacodynamics interactions. More recently, Shafer and colleagues wrote two landmark papers [72, 73] on the analysis of pharmacodynamic drug interactions in anesthesia. These authors explored the analysis of the interaction of drugs that compete for a single receptor and postulated that agents that act at a similar receptor must be additive in their combined effects. This is why inhalational anesthetic agents interact in an additive manner. In contrast, the authors state that synergy implies multiple sites of action by definition and that additivity among drugs acting on different receptors is only likely if the concentrations responsible for the drug effect of interest are well below the concentration associated with 50 % receptor occupancy.
Four types of pharmacodynamic interaction may be distinguished. Zero interaction, or additivity, is said to occur when a combination exerts an effect that is exactly the sum of the action of the individual agents. Combinations of inhalational anesthetics generally exert an additive interaction. The combination of 0.5 MAC halothane and 0.5 MAC sevoflurane exerts an effect of 1 MAC. Supra-additivity, synergism, or potentiation is said to occur when a combination of drugs exerts an effect that is stronger than the sum of the action of the individual agents. One then needs relatively less of the combination compared to the use of the individual agents. The interaction between inhalational agents and opioids, and between intravenous hypnotic agents and opioids, is generally synergistic in nature. Infra-additivity is said to occur when a combination exerts less effect than the sum of the action of the individual agents. Lastly, antagonism takes place when the effect of the combination is less than that of the action of one of its constituents [74].
Pharmacodynamic Interaction Between Intravenous Hypnotics and Opioids
Pharmacodynamic interactions between agents given intraoperatively may affect the pharmacodynamic profile of the combination considerably. Opioids exhibit strong analgesic effects, but also cause sedation at high drug concentration. Intravenous hypnotic agents, like propofol, induce and maintain surgical hypnosis but at a high concentration also suppress nociception. It is therefore no surprise that intravenous hypnotics and opioids support each other’s action in safeguarding general anesthesia. Propofol diminishes the opioid requirements needed for suppression of nociception, whereas opioids diminish the propofol concentration that is needed for induction and maintenance of surgical hypnosis. The interaction between intravenous hypnotics and intravenous opioids generally is found to be synergistic [5]. Alfentanil and fentanyl affect the blood propofol concentration needed to induce loss of consciousness in a synergistic manner [75, 76]. The EC50 for loss of consciousness with propofol is reduced by 50 % from 3.6 μg/ml to 1.8 μg/ml in the presence of a plasma alfentanil concentration of 300 ng/ml. The hemodynamic depression of the combination of propofol and alfentanil is more or less similar compared to when propofol is administered alone to induce loss of consciousness. The blood propofol concentration needed for loss of consciousness could not be reduced below 1.2 μg/ml, no matter how high the alfentanil concentration, exhibiting a ceiling in the hypnotic sparing effect by alfentanil .
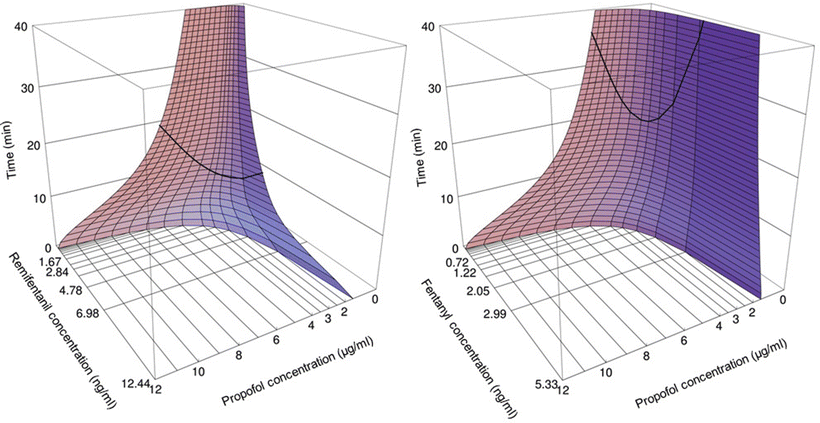
This is different for the maintenance of anesthesia. With a fivefold increase in the blood propofol concentration from 2 to 10 μg/ml, the alfentanil requirements are reduced by over tenfold. Alfentanil at high concentrations postpones recovery. In the presence of a significant plasma alfentanil concentration of 150 ng/ml, the blood propofol concentration has to decrease to as low as 0.5–1 μg/ml before patients regain consciousness [4, 5, 8, 9, 77–81]. In contrast, in the presence of a plasma alfentanil concentration below 50 ng/ml, patients are already awake postoperatively at blood propofol concentrations of 2–3 μg/ml. For remifentanil [82] and propofol, the interaction for intraoperative end points and awakening run parallel to those between alfentanil and propofol (Fig. 17.3) [83– 85]. Fentanyl and propofol also interact in this synergistic manner [76, 86, 87]. One may conclude that propofol concentrations at which patients regain consciousness are affected by the degree of painful stimulation at awakening postoperatively and by concurrent opioid concentration. The propofol EC50 reduction for intraoperative anesthetic stability by alfentanil and remifentanil is similar with a potency ratio of alfentanil /remifentanil of 35:1 [5].
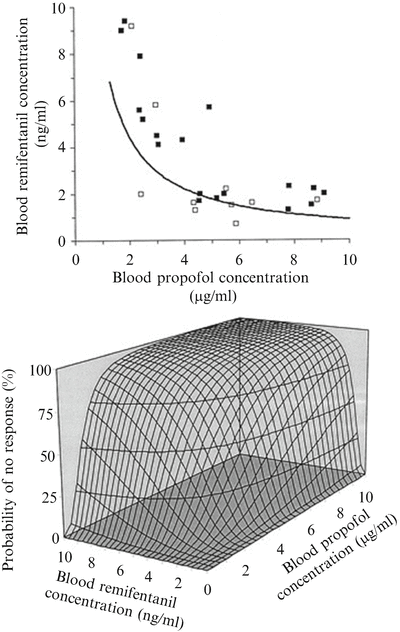
Fig. 17.3
Concentration–effect relation of the combination of propofol and remifentanil for suppression of responses to intubation. The iso-effect curve (=isobole) in the upper graph was obtained by response surface modeling of the response (open squares)–no response (closed squares) data on intubation versus the corresponding measured blood propofol concentrations and blood remifentanil concentrations. The displayed iso-effect curve represents remifentanil and propofol concentrations associated with a 50 % probability of no response to intubation, describing the synergistic interaction model. In the concentration–response surface (bottom) for the combination of propofol and remifentanil, the isoboles for 25, 50, and 75 % probability of no response are shown (Adapted from Mertens et al. [83] with permission from Wolters Kluwer Health, Inc.)
By means of computer simulations, based both on pharmacokinetic and pharmacodynamic interaction data, the optimal propofol–opioid concentration combination has been defined that assures both adequate anesthesia and the most rapid possible recovery in 50 % of patients [4]. This optimal propofol–alfentanil concentration combination has been determined to be a blood propofol concentration of 3.5 μg/ml in the presence of 85 ng/ml of alfentanil . The optimal propofol concentration is much lower when combined with remifentanil compared to when combined with fentanyl, sufentanil, or alfentanil [4]. Whereas the optimal propofol concentration (EC95) when combined with fentanyl is of the order of 5 μg/ml, the optimal propofol concentration is 2.5 μg/ml in the presence of remifentanil (Fig. 17.4). From the optimal propofol–opioid concentrations, optimal propofol and opioid infusion schemes have been derived that assure adequate anesthesia and the most rapid return of consciousness after termination of the infusion when propofol is combined with one of the opioids fentanyl, alfentanil , sufentanil, or remifentanil (Table 17.2). These infusion schemes should be used as guidelines, and adjustments should be made to the individual needs of the patient in anticipation of factors such as age, gender, and stimulus intensity related to the type of surgery. The overall message, though, remains clear; the propofol–opioid interaction is synergistic and the optimal propofol infusion regimen is different for the various opioids .