INTRODUCTION
The medicinal benefits of cardiac glycosides have been recognized for centuries, and even with other alternative medications, digitalis preparations, such as digoxin, are still used for the treatment of atrial fibrillation and symptomatic congestive heart failure.1 In addition to availability as pharmaceuticals, cardiac glycosides are also found in plants such as foxglove, oleander, red squill, and lily of the valley. Similar cardioactive steroids are also found in the skin of toads in the Bufonidae family and in some herbal medications. Despite declining use of digoxin, the prevalence of patients diagnosed with digoxin toxicity has remained constant, and the use of digoxin-specific antibody fragments has increased.2 Digitoxin, a cardiac glycoside similar in structure to digoxin but with a longer half-life, is no longer commercially available in the United States, but is available in Canada and elsewhere in the world.
PATHOPHYSIOLOGY
Digoxin is a cardiac glycoside available for oral or intravenous use. Following oral absorption, digoxin reaches a maximal serum concentration 1 to 3 hours after ingestion. It is approximately 25% protein bound and has a large volume of distribution (6 to 7 L/kg). The drug is primarily eliminated through the kidneys.
Digoxin, like other cardiac glycosides, inhibits sodium-potassium ATPase.3 This inhibition results in increased intracellular sodium and increased extracellular potassium. As a result of the increased intracellular sodium, the sodium-calcium antiporter is not able to effectively remove calcium from the myocyte. Consequently, there is an increase in intracellular calcium, which augments inotropy. The increased intracellular calcium can contribute to delayed after-depolarizations, which may lead to premature ventricular contractions and dysrhythmias. In addition, there is a decreased refractory period of the myocardium, which increases automaticity and hence is associated with an increased risk of dysrhythmias. Furthermore, cardiac glycosides shorten atrial and ventricular repolarization, thereby decreasing the refractory period and thus increasing automaticity.
Cardiac glycosides also increase vagal tone via action at the carotid body, thereby reducing conduction through the sinoatrial and atrioventricular nodes. In toxic concentrations, cardiac glycosides can increase sympathetic tone. Digoxin can reduce plasma renin concentrations in patients with advanced heart failure, thereby resulting in peripheral vasodilation.4 In contrast, in those without heart failure, digoxin can cause vasoconstriction. This difference is likely due to increased sensitivity of the carotid baroreceptors in patients with advanced, chronic heart failure.5
CLINICAL FEATURES
Digoxin has a narrow therapeutic index, and toxicity results from an exaggeration of its pharmacologic activity. The timing and clinical presentation of acute versus chronic digoxin toxicity differ significantly (Table 193-1).6 In addition to cardiac manifestations such as syncope and dysrhythmia, digoxin toxicity may present with GI distress, dizziness, headache, weakness, malaise, delirium, or confusion. Thus, an elderly patient taking digoxin who presents with mental status changes should be evaluated for toxicity.
| Acute Toxicity | Chronic Toxicity | |
|---|---|---|
| Clinical history | Intentional or accidental ingestion | Typically elderly cardiac patients taking diuretics; may have renal insufficiency |
| GI effects | Nausea and vomiting, abdominal pain, anorexia | Nausea, vomiting, diarrhea, abdominal pain |
| CNS effects | Headache, dizziness, confusion, coma | Fatigue, weakness, confusion, delirium, and coma are often prominent features |
| Cardiac effects | Bradydysrhythmias or supraventricular tachydysrhythmias with atrioventricular block | Almost any ventricular or supraventricular dysrhythmia can occur; ventricular dysrhythmias are common |
| Electrolyte abnormalities | Hyperkalemia | Normal, decreased, or increased serum potassium, hypomagnesemia |
| Digoxin level | Marked elevation (if obtained within 6 h) | Minimally elevated or within “therapeutic” range |
Patients with acute digoxin toxicity tend to have more abrupt onset of symptoms than those with chronic toxicity. In acute cardiac glycoside poisoning, there may be an asymptomatic period of several hours before the onset of symptoms. GI symptoms, such as nausea, vomiting, anorexia, and vague abdominal pain, are often the earliest manifestations of acute toxicity. Increased central vagal tone typically produces cardiac manifestations such as bradydysrhythmias or atrioventricular block. Neurologic manifestations such as weakness or confusion can occur independently of the blood pressure. The classic description of digoxin toxicity includes viewing yellow-green halos around objects, termed xanthopsia. However, patients more frequently describe nonspecific changes in their color vision.7
Hyperkalemia is an important finding in acute toxicity and may develop due to inhibition of sodium-potassium ATPase. Digoxin levels obtained with the first 6 hours following an acute ingestion may be falsely elevated as the level represents a predistribution level rather than reflecting the amount ingested. Overall, the severity of acute toxicity correlates most closely with the degree of hyperkalemia and correlates poorly with the early serum digoxin levels.8
Chronic toxicity occurs most typically in the elderly and is often the result of drug–drug interactions or declining renal function. Some of the more common drug interactions that predispose to chronic digoxin toxicity include calcium channel antagonists, amiodarone, β-receptor antagonists, diuretics, indomethacin, clarithromycin, quinidine, procainamide, and erythromycin. In particular, interaction between digoxin and clarithromycin contributes to increased hospitalizations for digoxin toxicity in elderly patients.9 A common scenario involves a patient starting to take a diuretic, which results in mild dehydration and hypokalemia; dehydration reduces the clearance of digoxin, and hypokalemia increases susceptibility to digoxin, resulting in chronic toxicity.
Decreases in renal function and lean body mass associated with aging may alter the pharmacokinetics of digoxin, leading to toxicity at normally therapeutic doses.10 This population may also possess a higher risk due to coexisting diseases and polypharmacy.10,11
The patient with chronic digoxin toxicity often has vague and nonspecific signs and symptoms compared to the patient with acute digoxin toxicity. GI symptoms may occur but may be less pronounced. Neurologic manifestations, such as weakness, fatigue, confusion, or delirium, are more prominent features in chronic toxicity.6,12
DIAGNOSIS
The diagnosis of digoxin toxicity is a composite picture, using history, physical examination, and laboratory studies; no single element excludes or confirms the diagnosis. In patients with heart failure and normal renal function, daily digoxin doses are usually between 125 and 250 micrograms. Digoxin toxicity can occur with a single ingestion of 1 to 2 milligrams in an adult, and fatalities have been reported following an acute ingestion of 10 milligrams in an adult and 4 milligrams in a child.
Differential diagnosis includes other toxins that may induce bradydysrhythmias such as calcium channel antagonists, β-receptor antagonists, class IA antidysrhythmics (procainamide and quinidine), class IC antidysrhythmics (flecainide and encainide), clonidine and other imidazolines, and organophosphate or carbamate insecticide poisoning. Glycoside-containing and other cardiotoxic plants should also be considered (e.g., foxglove, squill, lily of the valley, oleander, rhododendron, monkshood, tobacco, false hellebore, and yew berry). Sick sinus syndrome, with its combination of supraventricular dysrhythmias and cardiac conduction blocks, can also mimic digoxin toxicity. Hyperkalemia from any cause may produce bradycardia and abnormal cardiac conduction and should be considered in the differential diagnosis.
Almost any cardiac dysrhythmia may be observed in digoxin toxicity, with the exception of rapidly conducted atrial dysrhythmias.13,14 The most common dysrhythmia in digoxin toxicity is premature ventricular contractions. Ventricular dysrhythmias occur more frequently in chronic than in acute poisonings. Although rare and not pathognomonic for digoxin toxicity, bidirectional ventricular tachycardia should be investigated for possible toxicity because only a few xenobiotics are known to produce this unique dysrhythmia, digoxin included.
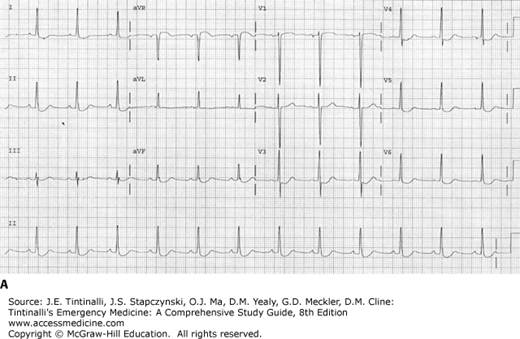
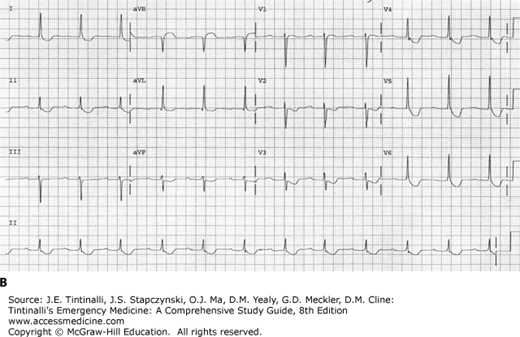
Four specific electrocardiographic findings are seen with therapeutic levels of digoxin and are not indicators of toxicity, although they may also be seen in poisoned patients. These findings include T-wave changes such as flattening or inversion, QT-interval shortening, a “scooped” appearance of the ST segment with ST-segment depression, and an increase in U-wave amplitude (Figure 193-1).14
FIGURE 193-1.
ECG demonstrating findings seen with digoxin use. A. ECG shows scooping of ST segments and small U waves with a serum digoxin level of 0.9 nanogram/mL (1.15 nmol/L). B. ECG shows scooping of ST segments, flattening of T waves, and first-degree atrioventricular block with a serum digoxin level of 1.2 nanograms/mL (1.54 nmol/L).