INTRODUCTION AND EPIDEMIOLOGY
Chronic obstructive pulmonary disease (COPD) is characterized by persistent airflow limitation that is generally progressive and associated with an abnormal inflammatory response to noxious particles or gases.1,2,3,4,5,6 COPD has two main forms: chronic bronchitis, defined in clinical terms, and emphysema, defined in terms of anatomic pathology. This traditional categorization is often indistinct, limiting the clinical utility of the definitions.2,3,4,5,6 Chronic bronchitis is the presence of chronic productive cough for 3 months in each of 2 successive years, where other causes of chronic cough have been excluded.2,3,4,5,6 Emphysema results from destruction of bronchioles and alveoli. The World Health Organization’s Global Initiative for Chronic Obstructive Lung Disease definition of COPD encompasses chronic bronchitis, emphysema, bronchiectasis, and asthma, and acknowledges that most patients have a combination of the different diseases.
COPD accounted for 715,000 U.S. hospitalizations in 2010,7 with $49.9 billion estimated as the cost for care.7 The prevalence of COPD in women has doubled in the past few decades, and women now account for >50% of COPD-related deaths; the prevalence has remained stable in men.8 The prevalence of COPD is highest in those countries that have the greatest cigarette use.
CHRONICALLY COMPENSATED CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Although tobacco smoke is the major risk factor for developing COPD, only 15% of smokers will develop COPD. Occupational dust, chemical exposure, and air pollution are other risk factors for COPD. α1-Antitrypsin deficiency accounts for <1% of COPD patients.
Irritants, notably tobacco smoke and air pollutants, trigger an increase in inflammatory cells in the airways, lung interstitium, and alveoli. Proteases eventually break down lung parenchyma and stimulate mucus secretion. Mucus-secreting cells replace cells that normally secrete surfactant and protease inhibitors. These changes result in a loss of elastic recoil, narrowing, and collapse of the smaller airways. Mucous stasis and bacterial colonization develop in the bronchi. The earliest objective changes in the evolution of COPD are clinically imperceptible; these early changes are small increases in peripheral airway resistance or lung compliance. Because dyspnea and hypersecretion often progress insidiously, it may take decades before COPD becomes clinically evident. The Global Initiative for Chronic Obstructive Lung Disease guidelines are helpful for the early diagnosis and treatment of COPD (Table 70-1),6 although there is only a weak correlation between forced expiratory volume in 1 second (FEV1), symptoms, and health-related quality of life.6
The central element of chronic lower airway obstruction is impedance to expiratory airflow due to increased resistance or decreased caliber of the small bronchi and bronchioles. Airflow obstruction results from a combination of airway secretions, mucosal edema, bronchospasm, and bronchoconstriction. Exaggerated airway resistance reduces total minute ventilation and increases respiratory work.
In emphysema, alveolar and capillary surfaces are distorted or destroyed, resulting in alveolar hypoventilation and ventilation–perfusion mismatch. The result is hypoxemia and hypercarbia. Sleep may blunt the ventilatory response to hypercarbia. The right ventricle hypertrophies and dilates, resulting in pulmonary hypertension and right ventricular failure. Right ventricular pressure overload is associated with atrial and ventricular arrhythmias. (See chapters 57, “Systemic Hypertension and chapter 58, “Pulmonary Hypertension.”)
The hallmark symptoms are chronic and progressive dyspnea, cough, and sputum production; these may vary from day to day.2,3,4,5,6 Minor hemoptysis is frequent, especially in chronic bronchitis and bronchiectasis, although it may herald lung carcinoma. Physical findings may include tachypnea, accessory respiratory muscle use, or pursed-lip exhalation. Lower airway obstruction causes expiratory wheezing, especially during maximum forced exhalation, and prolongation of the expiratory time. Patients with chronic bronchitis exhibit coarse crackles as uncleared secretions move about the central airways. In patients with emphysematous disease, there is expansion of the thorax, impeded diaphragmatic motion, and global diminution of breath sounds. Poor dietary intake and excessive caloric expenditure for the work of breathing cause weight loss, notably in emphysema. In the early stages, arterial blood gas measurements reveal mild to moderate hypoxemia without hypercapnia.
As COPD advances, especially when the FEV1 falls below 1 L, hypoxemia becomes more severe and hypercapnia develops. Arterial oxygenation worsens during acute exacerbations, exercise, and sleep. Clinical signs of severe COPD include facial vascular engorgement from secondary polycythemia, and tremor, somnolence, and confusion from hypercarbia. Right heart failure may occur and be seen as edema or ascites, and the signs are often disguised or underestimated by the seemingly more overwhelming signs of respiratory disease. If concomitant left heart failure exists, the cardiac auscultatory findings may be overshadowed by the pulmonary inflation abnormalities of COPD.
The diagnosis of chronic, compensated COPD is confirmed by spirometry: a postbronchodilator FEV1 of <80% predicted, and a ratio of FEV1 to forced vital capacity of <0.7.6 Once the disease progresses, the percentage of predicted FEV1 is a better measure of disease severity.2,3,4,5,6
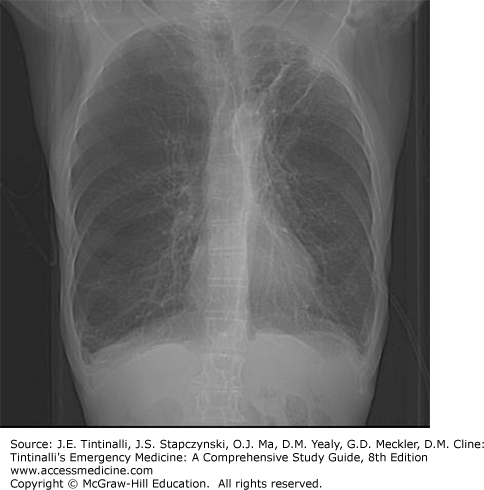
Chronic bronchitis is not radiographically apparent unless bronchiectasis is present. In emphysema, radiographs show hyperaeration, seen as increased anteroposterior chest diameter, flattened diaphragms, increased parenchymal lucency, and attenuation of pulmonary arterial vascular shadows (Figure 70-1).
Distinguishing acute heart failure from COPD is difficult. A B-type natriuretic peptide level <100 picograms/mL supports a diagnosis of COPD; levels >500 picograms/mL have a sensitivity of 80% and positive predictive value of 47% for acute heart failure (see chapter 62, “Respiratory Distress”).9 The ECG detects dysrhythmias or ischemia but does not accurately assess the severity of pulmonary hypertension or right ventricular dysfunction.
Treatment for chronic compensated COPD includes oxygen, pharmacotherapy, measures to decrease mucus secretion, smoking cessation, and pulmonary rehabilitation.
Long-term oxygen therapy reduces COPD mortality. The goal of long-term oxygen therapy is to increase the baseline partial pressure of arterial oxygen (PaO2) to ≥60 mm Hg or the arterial oxygen saturation (SaO2) to ≥90% at rest. Criteria for long-term oxygen therapy are a PaO2 ≤55 mm Hg, a SaO2 ≤88%, or a PaO2 between 56 and 59 mm Hg when pulmonary hypertension, cor pulmonale (sustained right ventricular failure), or polycythemia is present.6
Pharmacotherapy does not alter disease progression but provides symptomatic relief, controls exacerbations, improves quality of life, and improves exercise performance.10 Inhaled long-acting β2-agonists are preferred over short-acting formulations, coupled with anticholinergics. Combining bronchodilators with different mechanisms and duration of action may increase bronchodilation without increasing side effects.11 Combination inhalers of short-acting β2-agonists with anticholinergic agents include fenoterol/ipratropium and salbutamol/ipratropium. Long-acting inhaled β2-agonists, such as salmeterol, formoterol, olodaterol, and indacaterol, are used on a regular basis, adding short-acting inhaled β2-agonists, usually albuterol, as needed. Anticholinergic agents cause bronchodilation by blocking the effect of acetylcholine on muscarinic-3 receptors. Long-acting anticholinergic agents, such as tiotropium, aclidinium, and glycopyrronium, are preferred over short-acting agents, such as ipratropium bromide or oxitropium bromide.12,13 Bronchodilators often only chronically improve FEV1 by 10%.
Experts do not recommend long-term systemic corticosteroid therapy for all COPD patients,14 because only about 20% to 30% improve. Short-term steroid use (days) aids in treating exacerbations. Regular treatment with inhaled corticosteroids is indicated for patients with a documented spirometric response to inhaled corticosteroids, those with an FEV1 of <50%, or those with predicted and recurrent exacerbations requiring antibiotic treatment or systemic corticosteroids.6 Long-term treatment with inhaled corticosteroids added to long-acting bronchodilators is recommended for patients at high risk of exacerbation. Combination inhalers with long-acting β2-agonists plus corticosteroids include formoterol/budesonide, formoterol/mometasone, salmeterol/fluticasone, and vilanterol/fluticasone.6
Theophylline is relegated to an adjunct COPD therapy.15 Theophylline inhibits phosphodiesterase and has an anti-inflammatory effect. It is not commonly used, but can be used in some patients not well controlled with inhaled corticosteroids or long-acting β2-agonists. Although retrospective studies suggest that statins decrease the rate and severity of exacerbations, rate of hospitalization, and mortality, a large prospective trial failed to demonstrate benefit of daily simvastatin over placebo.16 Daily azithromycin may decrease acute exacerbations in older patients and those with milder Global Initiative for Chronic Obstructive Lung Disease staging.17
Respiratory secretions are kept mobilized by generous oral fluid intake and room humidification. Limit the use of antihistamines, antitussives, mucolytics, and decongestants. Expectorants are not of clear benefit.
Smoking cessation is the only intervention that can reduce both the rate of decline in lung function6 and mortality from respiratory causes.2,3,4,5,6 The ED is a site to attempt smoking cessation interventions.18 A combination of nicotine replacement therapy or medications and behavioral interventions can assist patients with smoking cessation, especially with referral to a program.19
Pulmonary rehabilitation can improve exercise capacity and quality of life and is recommended in patients with moderate to severe COPD. Pneumococcal vaccination and influenza vaccination are key to dampen acute infections.6
ACUTE EXACERBATIONS OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Acute exacerbations of COPD are characterized by worsening of respiratory symptoms beyond normal day-to-day variations20 and are usually triggered by an infection or respiratory irritant. More than 75% of patients with acute exacerbations have evidence of viral or bacterial infection, with up to half specifically due to bacteria.21,22 Other important triggers for exacerbations are hypoxia, cold weather,23 β-blockers, narcotics, or sedative-hypnotic agents. The final common pathway for an exacerbation is the release of inflammatory mediators that result in bronchoconstriction, pulmonary vasoconstriction, and mucus hypersecretion. The work of breathing increases due to higher airway resistance and lung hyperinflation. The oxygen demand of respiratory muscles increases, generating additional carbon dioxide and causing hypercapnia, resulting in further physiologic stress.23 Acute exacerbations of COPD are primarily due to ventilation–perfusion mismatch rather than the expiratory airflow limitation seen with asthma exacerbations.24 Supplemental oxygen increases blood oxygen concentrations and can help reverse pulmonary vasoconstriction.
The most life-threatening feature of an acute exacerbation is hypoxemia (arterial saturation <90%). Signs of hypoxemia include tachypnea, tachycardia, systemic hypertension, cyanosis, and a change in mental status. With increased work of breathing, carbon dioxide production increases; alveolar hypoventilation creates arterial carbon dioxide retention and respiratory acidosis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


