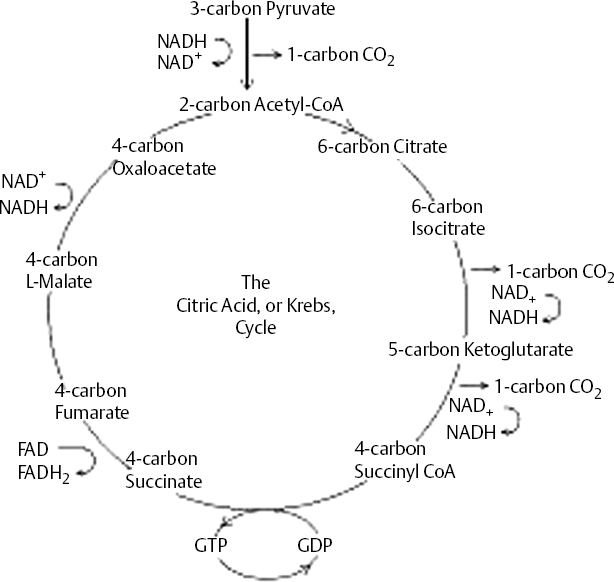
18 Dennis Cramer, Dan Miulli, and Javed Siddiqi The ultimate result of a severe head injury primarily depends on the extent of injury at the time of incident. A primary brain injury from trauma includes contusions, epidural or subdural hematomas, intracerebral hemorrhage, and diffuse injuries. Initially, surgical intervention for a space-occupying mass should always be considered before proceeding to the management of secondary brain insults. These secondary brain injuries include edema, ischemia, and hypoxemia, which can result in infarction and/or herniation. Upon secondary injury, the brain undergoes numerous pathological and chemical changes that significantly impact the patient’s neurologic outcome. The mainstay of treating a secondary brain insult is to ensure adequate cerebral blood flow (CBF) via control of intracranial and cerebral perfusion pressures. Because CBF is difficult to measure, physicians can easily follow the mean arterial and intracranial pressures to estimate cerebral perfusion pressure (CPP). CBF is related to mean arterial pressure (MAP) and ICP, as shown in these equations: CBF = CPP/CVR = (MAP – ICP)/CVR, where CVR is cerebrovascular resistance. The equations show the complex relationship between brain perfusion and blood pressure. The Monro-Kellie hypothesis, which helps to explain the significance of ICP, states that the sum of the intracranial volumes of blood, brain tissue, and cerebrospinal fluid (CSF) are constant and that any increase in one component will be counterbalanced by a decrease in another. The volume of brain tissue is 90%; blood and CSF are 5% each. All of these are contained within the rigid skull. The introduction of additional volume, such as from hemorrhage or edema, must be compensated by changes in the blood or CSF volumes. Failure to counterbalance these changes will result in increased ICP and possibly intracranial shifts (herniation). Increased ICP can be compensated by shifting CSF from the ventricles and subarachnoid space to the spinal canal or by decreasing the intracranial blood volume via collapsing veins and constricting cerebral arteries. Changes in vessel diameter can result in a reduction of cerebral intravascular volume by as much as 70 mL, easily buffering a new volume mass, such as a hemorrhage.1 After compensatory mechanisms for increased ICP are exhausted the patient begins suffering from secondary brain insults. Eighty to 90% of traumatic brain-injured patients who die are found to have histopathologic evidence of cerebral ischemia.2 Studies show that approximately one third of patients with traumatic brain damage experience an “ultra-early” period of significantly decreased cerebral blood within 6 hours of injury.3 The ischemia is characterized by focal reductions in CBF below the threshold of 18 mL/100 g/minute, resulting in ischemic neuronal cell death.4 The hypoxic conditions perpetuate a hypermetabolic rate, resulting in an aerobic to anaerobic metabolism and leading to an increased concentration of lactic acid.5 Ionic homeostasis is then compromised, leading to a complex process of calcium influx and cellular injury and death.6 An estimated 15% of all head-injured patients suffer from refractory ICP.7 When there is deteriorating CPP in the face of increasing ICP, hypertonic saline can be used to increase blood osmolarity, expand plasma volume, and reduce brain edema via the extraction of water from neurons and interstitial tissue. Hypertonic saline results in improved microvascular blood flow, thereby lowering ICP. Variable concentrations of hypertonic saline from 3 to 23.4% have been used. Although limited use of hypertonic saline is relatively safe, prolonged use can affect cellular homeostasis, and, in the alcoholic, rapid correction of hyponatremia can lead to central pontine myelinolysis. However, when this and all other medical and surgical modalities have failed to adequately control elevated ICP, the physician may attempt the use of pharmacologic agents to reduce cerebral metabolic rate (CMR) and hence ICP in a patient who is deemed salvageable. Many pharmacologic agents have been used to lower ICP, such as pentobarbital, thiopental, etomidate, propofol, isoflurane, and desflurane, but not enough data exist to recommend one drug over another. Most published information addresses the use of barbiturates, especially pentobarbital. In 1974, Shapiro et al were the first to report the use of pentobarbital-augmented hypothermia in reducing CMR and ICP.8 This therapy remains controversial mainly because of the potential side effects of myocardial hypotension, hypothermia, immunosuppression, hypokalemia, and hepatic and renal dysfunction.9 Accordingly, the Brain Trauma Foundation, in cooperation with the American Association of Neurological Surgeons, has recommended a guideline using high-dose barbiturate therapy in hemodynamically stable head-injured patients with elevated ICP resistant to maximal medical and surgical treatment modalities.9a Because of marked hypotension seen in ~50% of patients treated with barbiturates, many will require cardiac inotropes and/or vasopressors to maintain an MAP resulting in a CPP ≥ 70 mm Hg,10 unless increased secondary risk concerns favor an acceptable lower CPP. Dopamine (primarily a vasoconstrictor) is commonly used starting at 3 μg/kg/minute and titrating to a maximum 20 μg/kg/minute to maintain CPP ≥ 70 mm Hg. Phenylephrine starting between 100 and 180 μg/minute, with a maintenance dosage of 40 to 60 μg/minute, or Levophed at 8 to 12 μg/minute is frequently used for progressively decreasing CPP and/or hypotension. Swan-Ganz catheterization and hemodynamic monitoring may be necessary to adjust cardiac medications and to ensure adequate volume status. Many protocols have been used to administer barbiturate therapy. Eisenberg’s protocol starts with a loading dose of pentobarbital at 10 mg/kg intravenous (IV) over 60 minutes, followed by 5 mg/kg every hour for 3 hours, with a maintenance dosage of 1 to 3 mg/kg/hour.11 In a study by Cormio et al on 67 severely head-injured patients undergoing treatment for refractory intracranial hypertension, pentobarbital loading doses decreased ICP and MAPs on average by 12 and 9 mm Hg, respectively.12 It can generally be considered that if the induced pentobarbital coma does not lower the ICP within 1 to 4 hours, it is unlikely to succeed without any other therapy. Thiopental has been used when large doses of pentobarbital are not available or when a rapidly acting barbiturate is needed. It works by producing cerebral metabolic depression and cerebral vasoconstriction. The loading dose is 5 mg/kg IV over 10 minutes, with a continuous infusion of 5 mg/kg/hour (range: 3–5 mg) for 24 hours. After 24 hours, the infusion may be decreased to 2.5 mg/kg/hour because fat stores have now become saturated. Propofol is a well-established sedative-hypnotic agent that has been used as an alternative to control elevated ICP, but questions remain regarding the pharmacodynamics of the drug. A study by Oertel et al has suggested a key mechanism responsible for the metabolic suppressive effect is a decrease in CO2 production, resulting in a global “pharmacological hypocapnia.”13 The starting dose is 5 to 10 μg/kg/minute and increases by 5 to 10 μg/kg/minute every 5 to 10 minutes until ICP is controlled. Like the barbiturates, propofol exhibits hypotensive effects that may require cardiac medications or eventual discontinuation. In addition to the cardiotoxic effects, propofol may cause electrocardiogram changes and discoloration of urine. Pharmacologically induced burst suppression is generally maintained 3 to 7 days; if computed tomography (CT) does not show any new findings, and ICP has been controlled, then therapy can be withdrawn slowly by reducing the infusion rate while monitoring ICP. Because of prolonged high-dose barbiturate therapy and the long half-lives of these drugs, it is difficult to distinguish the residual pharmacologic effects from the clinical condition. If a high index of suspicion for brain death is entertained, a nuclear cerebral metabolic test is warranted; if the test is negative, one should proceed with a cerebral angiogram before pronouncing brain death. The prophylactic use of barbiturates for ICP treatment has shown no benefits in patients with intracranial mass lesions and was even found to be harmful in patients with diffuse injury (mortality of 77% vs 41% in the mannitol group).14 A 2000 Cochrane Database review evaluating barbiturate therapy in acute traumatic brain injury concluded that there was no evidence that barbiturate therapy improves outcome for patients with acute severe brain injuries. The review also demonstrated that barbiturate therapy results in a decrease in blood pressure in 1 in 4 patients, and this hypotensive effect will offset any ICP-lowering effect on cerebral perfusion.14 Alternative agents to pentobarbital have not been well studied, but they have been used because of easier accessibility. A study comparing etomidate, isoflurane, and thiopental in animals undergoing 3 hours of middle cerebral artery occlusion found injured brain volume largest in the etomi-date and isoflurane groups and smallest in the thiopental group.15 Control of temperature is of extreme importance in the head-injured patient. There have been many small studies on the effect of hypothermia causing reduced cerebral metabolism and ICP. In 1993, Clifton et al reported a 16% increase in favorable outcome in head-injured patients who underwent induced hypothermia (32–33°C).16 A small study from 1999 showed mild hypothermia (34°C) to be an effective ICP control in patients with focal cerebral lesions.17 Tokutomi and colleagues reported that lowering body temperatures to 35 to 36°C could reduce intracranial hypertension while maintaining CPP without significant cardiac dysfunction or oxygen delivery abnormalities.18 However, the results from a prospective, multicenter, randomized study consisting of 392 subjects demonstrated that traumatic brain-injured subjects who were randomly assigned to treatment with hypothermia (33°C) initiated within 6 hours of incident and maintained for 48 hours or nor-mothermia showed no difference to normothermic patients at 6 months as measured by the Glasgow Outcome Score (GOS). Conversely, there were poorer outcomes in patients over 45 years old who were treated with hypothermia compared with the normothermia group. Also, the hypothermia group showed a higher cumulative fluid balance, greater use of vasopressors, and a higher percentage of days with complications.19 This well-conducted study brings a renewed interest in the field of hypothermic treatment and the need for future trials to elucidate a clearer understanding and treatment guidelines. Traumatic brain injury leads to numerous neurochemical events and is associated with a catecholamine stress response, including hyperglycemia.20 Many studies have shown hyperglycemia to correlate with the severity of brain injury and to be a predictor of neurologic outcome.21–23 A study by Young et al showed that patients who had the highest peak admission 24 hour serum glucose level had the worst neurologic outcome at 18 days from injury.21 The researchers also found that patients with a peak 24 hour serum glucose level >200 mg/dL had a 2 point decrease in their GOS and that those patients with a serum glucose level <200 mg/dL experienced a 4 point increase during the 18 day study period. Rovlias and Kotsou also found serum glucose levels >200 mg/dL in the first 24 hours to be highly predictive of an unfavorable outcome.24 Kushner et al found the median initial serum glucose level in acute ischemic cerebral infarction patients to be 155 mg/dL23 Additionally, clinical recovery was significantly poorer in patients with an initial glucose level greater than the median. The researchers reported that serum glucose admission levels correlated with the extent of metabolic brain abnormalities seen on acute positron emission tomography (PET) scanning. During normal aerobic metabolism, energy in the brain is formed primarily when glucose is oxidized to CO2 and water. The cellular metabolism of glucose begins with the process of glycolysis, resulting in the generation of pyruvate, the reduced form of nicotinamide-adenine dinucleotide (NADH), and adenosine triphosphate (ATP): Glucose + 2 ADP + 2 Pi + 2 NAD+ → 2 Pyruvate + 2 ATP + 2 NADH + 2 H+ + 2 H2O, where ADP is adenosine diphosphate. Pyruvate can be converted to lactate or to the amino acid alanine, or it can enter the citric acid, or Krebs, cycle (Fig. 18–1). The energy products derived from the citric acid, or Krebs, cycle (tricarboxyclic acid) are used in the final step of glucose metabolism as it enters the electron transport chain (ETC; Fig. 18–2). The end result of glucose breakdown is 38 moles of ATP, which is more efficient than the 6 moles of ATP produced via glycolysis alone. The detrimental effects of hypoxic or ischemic brain injury result from anaerobic metabolism in which glucose is converted to lactate and hydrogen ions, rather than pyruvate. With increased levels of glucose available for lactate production, excessive lactate accumulation leads to greater tissue acidosis and subsequently neuronal damage.25 There is considerable evidence of traumatic brain-injured patients with serum glucose levels correlated with neurologic outcome. However, further clinical trials are needed to address neurologic outcome with strict glucose control in the acute brain injury.
Cerebral Protection Measures
 Pharmacologic Cerebral Metabolic Depressants
Pharmacologic Cerebral Metabolic Depressants
 Hypothermia
Hypothermia
 Blood Glucose Control
Blood Glucose Control
![]()
Stay updated, free articles. Join our Telegram channel





Full access? Get Clinical Tree


