15 Dan Miulli and Javed Siddiqi The brain capillary varies from the systemic capillary by several unique features; these establish the blood–brain barrier. The brain capillary endothelial cells are the distinguishing elements of the blood–brain barrier. The brain capillary endothelial cells are not fenestrated, lack intracellular clefts, and are closed by intercellular tight gap junctions. The tight gap junctions of the brain capillary endothelial cells assist in keeping substances out of the brain interstitial space. The body systemic capillary endothelial cells are not in contact with one another; they do not make a circumferential ring barrier but instead have multiple fenestrations that allow fluid to pass freely. The circumferential ring of brain capillary endothelial cells regulates which substances reach the brain’s interstitial space, the supporting astrocytes, and the neurons. In an intact blood–brain barrier, all substances pass through the capillary endothelial cell and the other regulator, the choroid plexus epithelial cell. The blood–brain barrier regulates the biochemical, immunological, and electrical passage of material into the brain. The tight gap junctions, the strong intercellular connections, have a high electrical impedance, preventing ions from passing. The capillary endothelial cells exclude plasma proteins from the brain interstitial space because the endothelial cells have very little pinocytic vesicles to transport the proteins and contain unique cerebral enzymes, cellular channels, and transport systems. The brain capillary endothelial cells require large amounts of energy to serve as the complex regulatory interface between the brain and the blood circulation. The brain capillary endothelial cell energy dependent transport organizations have 3 to 5 times the amount of mitochondria as the systemic capillary cells. Maintaining the blood–brain barrier’s unique functions, the brain capillary membrane has a different structural surface, and its lack of permeability is assisted by pericytes and astrocytic foot process. The pericyte or perivascular cells are not unique to the brain; they are found partially wrapping systemic capillaries and venules in endothelial cells. The selective permissiveness into the brain interstitial space is not present in the neural hypophysis, subfornicele organ, median eminence, area postrema, and pineal gland (Table 15–1).1,2 In the normal state, for substances to pass from the blood to the brain interstitial space, they must pass through the capillary endothelial cell. The brain capillary endothelial cell allows certain compounds to pass with relative ease into the brain interstitial space via transcellular diffusion or ubiquitous transport mechanisms. These substances are lipid soluble, low molecular weight of ~ 500 daltons or less, nonpolarized, and not bound to proteins. Such substances include glucose, low-density lipoproteins, transferrin, bromide, morphine, and bile salts. These substances when not actively transported must still diffuse down a concentration gradient from blood to capillary endothelial cell to brain interstitial space. The blood–brain barrier is not permeable to ions and amino acids, and the exact type of permeability to water is controversial. Water does not enter through a lipid membrane but must pass through selective water channels regulated by aquaporins. The blood–brain barrier excludes water-soluble molecules with a molecular weight greater than 180 daltons. It is not permeable to vital dyes, epinephrine, curare, bile pigments, and fluorescein. Substances that enter the brain interstitial space fluid could alter nervous system function. These substances pass from the blood–brain capillary to the interstitial space, where they may be absorbed by neurons, glial cells, or others; they then pass through, but sometimes around, subependymal glial cells and ependymal gap junctions into the cerebrospinal fluid (CSF) of the ventricles, cisterns, and Virchow-Robin spaces, where they eventually circulate back into the blood via the arachnoid villi.1–3
Cerebral Perfusion
 Blood-Brain Barrier
Blood-Brain Barrier
Brain Interstitial Circulation
| Tight gap junction | Different membrane structure |
| Basement membrane | Neuronal enzymes |
| Minimal pinocytic activity | Transport mechanisms |
| High mitochondrial content | Pericytes (also seen in systemic circulation) |
| High electrical impedance | Astrocytic foot process |
Blood–Cerebrospinal Fluid Barrier
The choroid plexus epithelial circumferential junction actively secretes substances into the CSF. The blood–CSF barrier is similar to the blood–brain barrier of the capillary endothelial cell. Substances dissolved in the choroid capillary blood pass more easily through the choroid endothelium, are selectively passed by the connective tissue of the choroid plexus, and then are regulated by the choroid epithelium. The choroid epithelial cells contain circumferential junctions, which must secrete CSF into the ventricle. The CSF acts as a sink for the brain interstitial fluid, with a gradient of substances carrying components away from the interstitial space.
Blood–Brain Barrier Disruption
Diseases of the nervous system disrupt the blood–brain barrier, resulting in most of the devastation. The barrier is disrupted by hypertension, hypercapnia, hypocapnia, trauma, hyperthermia, chemicals such as intra-arterial lactated Ringer’s, mannitol, leukotriene C4, nitric oxide synthetase, arabinose, and lactamide, as well as in the center of tumors, but necessarily in the periphery. The disrupted blood–brain barrier leads to brain edema, accumulation of fluids and electrolytes, increased cerebral volume, increased intracranial pressure (ICP), cellular swelling, and cell death. There are many studies of brain edema in a variety of pathologies. However, there is a clinical situation that, when thoroughly understood, will clarify most questions about the blood–brain barrier and cerebral edema. That situation is head trauma.
Vasogenic Edema
At the moment of impact, the blood–brain barrier’s tight gap junctions are stretched, which essentially turns the brain capillary for ~30 to 60 minutes into a sieve, leading to vasogenic edema. There is accumulation of serum, albumin, and proteins. The opening is also prolonged for an unknown duration by hypoxia and for a single time of 6 hours by a hypertension surge. The mechanical disruption traumatizes microvessels, cells, and cellular organelles, releasing calcium, ornithine decarboxylase, and free radicals. Sustained increases of free radicals meant to oxidize toxins will disrupt the cellular membrane. An increase in intracellular and extracellular stores of calcium will be sustained by hypoxia.
The activity of ornithine decarboxylase is increased 2000% due to upregulation of its messenger ribonucleic acid (mRNA).4 It acts to decarboxylate ornithine forming putrescine, which is then converted into spermidine, then finally into spermine. All three are released immediately after cellular injury because of the necessity for regeneration, development, cell growth, and neuronal survival. Putrescine accumulation induces shrinkage in cerebral microvessels after trauma, opening the blood–brain barrier and increasing vasogenic edema. Putrescine can be increased by isoflurane and decreased by ketamine.
Bradykinins are released, which, with other pathway products, increase calcium at the time of mechanical disruption. This calcium increases calcium-calmodulin and calcium protein kinase C and opens the blood–brain barrier.
The disrupted blood–brain barrier is stabilized by dexamethasone, copper, zinc, superoxide dismutase, serotonin antibodies, and progesterone.
Cytotoxic Edema
The most common type of edema after head injury, cytotoxic edema, occurs in the cell 30 to 60 minutes after insult as a response to the intracellular accumulation of the toxic levels of lactate, hydrogen-ion acidosis, potassium, oxygen free radicals, and glutamate, among others. The sodium potassium adenosinetriphosphatase (ATPase) pump fails secondary to depletion of cellular energy, allowing sodium to build up intracellularly and drawing in water. Hydrogen accumulation is the most common reason for buffering the cell by increasing the water content. The cell osmotically swells, reacting to the increasing glycogen granules that are sequestered to provide more substrate to the tricarboxylic acid (TCA) cycle. The additional energy is required to supply cellular active transport mechanisms, which include
- Sodium potassium ATPase
- Sodium potassium chloride cotransporter
- Calcium-activated potassium channels
- Sodium hydrogen antiporter
- Chloride bicarbonate exchanger
- Carrier-mediated transport of nutrients
- Receptor-mediated transport of peptides
As the cellular membrane starts to fail, glutamate is released, increasing cellular metabolism, which, under its current anaerobic conditions, leads to further lactate production and, without utilization, accumulation and then to failure of energy metabolism and cell death.3
Interstitial Edema
Interstitial fluid drains down the gradient into the CSF sink. That sink begins to get clogged with toxins and proteins. CSF production is increased to buffer the brain interstitial fluid acidosis run off by up-regulation of the chloride bicarbonate exchanger. One method to decrease interstitial edema and the major contributor to traumatic brain injury cytotoxic edema is by decreasing the sink into which brain fluid drains. Decreasing the CSF in the ventricles does this; decreasing the pressure of the intracranial contents does not.
| Type of edema | Means to decrease |
| Cytotoxic (the most common type in head injury) | Alpha-difluoromethylornithine, α-trinositol, amiloride, THAM |
| Vasogenic | CuZn SOD, 5HT1B, progesterone, indomethacin, naloxone, dexamethasone, CSF drainage |
| Interstitial | CSF drainage, acetazolamide |
CSF, cerebrospinal fluid; CuZn SOD, copper-zinc superoxide dismutase; THAM, tris-(hydroxymethyl)-aminomethane.
There is no silver bullet to treat brain injury. Fortunately, the brain can adapt and recover from multiple insults. Brain injury occurs because several innate brain systems fail to compensate for the overwhelming insult. Currently, there are experimental chemicals that are available to ameliorate different types of edema (Table 15–2).5
The aim of aggressive early treatment is to prevent ischemia. Glucose and glycogen are depleted in 4 minutes; there is increased metabolism in the layer surrounding the ischemic penumbra, the brain temperature increases, metabolism increases, and brain tissue pO2 drops to less than 20 mm Hg, favoring anaerobic respiration. Anaerobic respiration causes lactic acidosis and brings hydrogen into the cell. Lactate accumulation approximates glucose stores at the time of ischemia. The cell swells in response to increasing levels of hydrogen. Multiple neuromodulators stimulate cyclic adenosine monophsphate (cAMP)–dependent ion transport, bringing more hydrogen into the cell. The ischemia leads to the cellular pH dropping to 6.0 to 6.4, inhibiting mitochondrial respiration and the ability to sequester calcium. When the cell is deprived of energy and oxygen, calcium stores are released, stimulating phosphorylation of protein kinase C and further increasing intracellular hydrogen in a vicious cycle, leading to degradation of cell membranes. Therefore, increasing cerebral blood flow in excess of edema is necessary to increase oxygen delivery.
 Cerebral Blood Flow
Cerebral Blood Flow
Cerebral blood flow (CBF) averages 45 to 65 mL/100 g/minute (55 mL/100 g/minute used by most references), being higher in gray matter (75–80 mL/100 g/minute) and somewhat less in white matter (20–30 mL/100 g/minute). Changes occur sooner when the entire CBF is affected compared to changes in regional CBF. Protein synthesis declines at 40 to 50 mL/100g/minute, and anaerobic glycolysis at 35 mL/100 g/minute. Drastic electroencephalography (EEG) changes of absent activity are seen as early as 25 mL/100 g/minute; however, brainstem auditory evoked responses (BAERs) occur at 12 mL/100 g/minute, and loss of transcellular function begins at 10 to 20 mL/100 g/minute and anoxic depolarization at 8 to 15 mL/100 g/minute. Brain tissue tolerates slightly lower regional CBF changes before similar effects. Of course, the duration of decreased perfusion also differentiates ischemia from infarction.6
Cerebral Vascular Resistance
The physiology of cerebral vascular resistance (CVR) differs from the systemic circulation. One half of the systemic mean arterial pressure (MAP) is lost just distal to the cerebral arteries and arterioles. CVR is complex and neither always proportional nor linear to cerebral perfusion pressure (CPP). Therefore, knowing CPP does not allow direct calculation of CBF. CVR varies with edema, PaCO2, and a host of other conditions. Under normal steady state conditions, autoregulation occurs as CVR varies with CPP to maintain a constant CBF. However, during extremes of pathological states, or extremes of autoregulation, CVR cannot vary enough. Between a CPP of 50 and 125 mm Hg, the blood vessel dilates and constricts to maintain constant CBF. At CPP of 50 to 60 mm Hg, the blood vessel is maximally dilated, and further decrease in CPP only results in decreased CBF. Likewise, at a CPP of 125 mm Hg, the blood vessel is maximally constricted. Any further increase in CPP is similar to increasing the flow through a rigid tube. This will cause vasogenic edema as the tight gap junctions are split apart. The amount of autoregulation or the amount of CVR changes daily, hourly, and by the minute. During head trauma, unless secondary injury occurs, or hypotension, ischemia, or hyperventilation, autoregulation is maintained but shifted to the left.7 To date there is no direct way to continuously measure CVR. Criteria are being developed utilizing transcranial Doppler ultrasound, its pulsatile index and resistance, in an attempt to quantify CVR and CBF, as given here:
CBF = CPP/CVR = MAP – ICP/CVR
Hypotension
After severe head injury around contusions, in punctate hemorrhages CBF cannot be maintained. CBF drops to less than 20 mL/100 g/minute and remains less than 30 mL/100 g/minute during the first 8 to 24 hours. It remains lowest under subdural hematomas and diffuse axonal injuries and, as expected, with hypotension. However, it is not only low around the area of injury; globally, it can be decreased to half the normal level after injury. A single episode of hypotension doubles the morbidity and mortality associated with severe head injury. Hypotension leads to vasodilation 165% above baseline, increasing cerebral blood volume and ICP. Hypotension is not used initially to treat severe head injury. Although hypertension to maintain CPP in the normal range is beneficial,8 if CPP exceeds the upper limit of autoregulation, whether artificially changed or not, it too will increase cerebral blood volume and ICP. Guidelines for the Management of Severe Head Injury,7 published by The Brain Trauma Foundation, recommends the following:
Guideline: Hypotension (SBP < 90 mm Hg) or hypoxia (apnea or cyanosis in the field, PaO2 < 60 mm Hg, saturation < 90%) must be scrupulously avoided, if possible, or corrected immediately
Option: Mean arterial pressures should be maintained above 90 mm Hg through the infusion of fluids throughout the patient’s course to attempt to maintain cerebral perfusion pressure greater than 70 mm Hg.
Systolic blood pressure and mean arterial pressure can be treated in the field. However, if there is severe head injury, the patient must be taken to a trauma center where a neurosurgeon is present. The neurosurgeon will insert an external ventricular drain (EVD) and ICP monitor.
Cerebral Perfusion Pressure
Older studies compared the CPP to mortality in the adult mostly male population and found that if the CPP was greater than 80 mm Hg the mortality at that time in history was only 35 to 40%. If the CPP was less than 80 mm Hg mortality increased for each 10 mm Hg decrease CPP yielding a mortality of 95% for a CPP less than 60 mm Hg. Other investigators also found that the single most important therapy was to keep the CPP greater than 60 mm Hg.9 Similarly, Guidelines for the Management of Severe Head Injury recommends that CPP should be maintained at a minimum of 70 mm Hg.7
Several caveats must be made. This is the recommendation for the average adult male, not for children and not for some adult females who have a normal systolic blood pressure (SBP), let alone MAP lower than 90 mm Hg. A CPP greater than 70 mm Hg should not be maintained at the expense of causing other injuries, such as pulmonary edema and acute respiratory distress syndrome (ARDS). Hlatky et al10 demonstrated that for maximum CPP treatment in patients with difficult-to-manage increased ICP, more fluids were used, more vasopressors were utilized, and there was a 5-fold increase in ARDS. If secondary injury is becoming apparent, and that secondary injury may decrease outcome in the management of severe head injury, then CPP should be kept above a minimum of 60 mm Hg.
When attempting to manage the patient with severe head injury, orders must be written to maintain the CPP above a certain level, such as 70 mm Hg, or a minimum of 60 mm Hg, and must be written to maintain the CPP below an upper limit. The upper limit is variable from 90 to 120 mm Hg.
Blood Pressure Management in Infarction
There is considerable debate regarding the appropriate blood pressure to maintain in a patient who has suffered ischemia or who has suffered an intracerebral hemorrhage (ICH). Lowering blood pressure extensively during cerebral ischemia can lead to infarction. Blood pressure usually increases during the first few hours after ischemia in an attempt to correct the underlying physiological problem; but once the injury has equilibrated, the blood pressure decreases within 24 hours, returning to the patient’s normal level in 4 to 7 days. The rise in blood pressure does not change CBF in areas of the brain where autoregulation is intact. However, in areas of ischemia where there is decreased blood flow, it is a requirement; coupled with dysfunctional autoregulation, there is a dramatic decrease in blood flow. There are no large randomized clinical studies in humans to support this theory. Current theories for treating elevated blood pressure in acute ischemic stroke are based largely on expert opinion and vary with regard to treatment thresholds. One 16-patient randomized study demonstrated no difference in outcome in patients who underwent antihypertensive treatment versus placebo. Several reports observed and recorded data from patients who presented with ischemia and attempted to correlate the data without experimental protocol. The researchers observed that for every 20 mm Hg drop in SBP, the relative risk of worsening within 36 hours of stroke onset increased by a factor of 0.66.11 There are several management schemes; one is to lower blood pressure by 15% over 24 hours. However, the current guidelines for management of acute post-stroke hypertension have not changed prescribing patterns. There still remains considerable disparity due to uncertainty caused by lack of evidence from randomized controlled trials.12
For patients with SBP >220 mm Hg or diastolic blood pressure (DBP) >120 to 140 mm Hg on two readings 5 to 10 minutes apart,
- Give labetalol 10 mg intravenously (IV) every 10 minutes up to 150 mg. If no response, use nitroprusside 0.5 to 10.0 μg/kg/minute.
Patients who have had a stroke and are hypertensive are at high risk for an additional stroke. Those patients should receive blood pressure treatment when elevated to 140/90 mm Hg or higher. However, the decision to treat blood pressure at these levels should be made 4 to 7 days after the return to baseline blood pressure was to be expected. Blood pressure should be gradually reduced to 120/80, the optimal level for reducing cardiovascular risk.11
Blood Pressure Management in Hemorrhage
Management of hemorrhagic stroke is controversial, and there is no standardized system for treatment. Some advocate lowering blood pressure to reduce the risk of bleeding, edema formation, and systemic hypertensive complications, whereas others advocate allowing blood pressure to run its natural course as a protective measure against cerebral ischemia. Current American Heart Association guidelines13,14 recommend maintaining MAP <130 mm Hg, SBP <180 mm Hg, and CPP >70 mm Hg. Lowering SBP to <160 mm Hg in the first hours after ICH may prevent additional bleeding. Ohwaki et al15 studied patients with ICH and hypertension. They lowered SBP to targets of 140, 150, or 160 mm Hg and followed hematoma enlargement, defined as an increase in volume of ≥ 140% or 12.5 cm.3 Hematoma enlargement occurred in 16 patients. Maximum SBP was significantly associated with hematoma enlargement (p = .0074). Maximum SBP was independently associated with hematoma enlargement (odds ratio per mm Hg: 1.04; 95% CI, 1.01–1.07). Target SBPs of ≥160 mm Hg were significantly associated with hematoma enlargement compared to those of ≤150 mm Hg (p = .025).
For hypertension treatment in acute ICH,
- For patients with a history of hypertension, maintain SBP <160 mm Hg.
- For patients with no history of hypertension, maintain SBP <150 mm Hg.
- Give labetalol 10 mg IV every 10 minutes up to 150 mg.
- If no response, give enalapril 0.625 mg IV q 6 hours; may repeat 0.625 mg q 16 minutes to a total of 2.5 mg q 6 hours.
There remains some debate in treating patients with ICH on whether surgery improves perfusion and thus outcome. The STICH trial randomized 1033 patients from 83 centers and reported that patients with spontaneous supratentorial ICH in neurosurgical units show no overall benefit from early surgery when compared to initial conservative treatment.16
There is new evidence that ICH progression can be minimized with recombinant activated factor VII (RFVII) and consequently reduce morbidity and mortality after ICH.17 One to three doses of 40, 80, or 160 μg/kg given within the first 3 to 4 hours after symptom onset, or in patients at risk of additional bleeding, such as those with coagulopathy, demonstrated a 3 month mortality of 18% when compared to the placebo group mortality of 29%.18
However, there still remain subsets of patients that may benefit from surgical evacuation of ICH. The risk for rebleed is highest in the first 3 to 6 hours and lowest after 12 hours. Waiting until the patient is herniating before surgical intervention is no better than medical management alone.
Patients who may benefit from surgery for ICH include
- Patients who are symptomatic from a cerebellar hemorrhage >3 cm or >30 cc
- Patients who have a supratentorial ICH >20 to 30 cc and who present with a Glasgow Coma Scale (GCS) score of 13–5 or a decrease of 2 GCS points (carefully weigh the decision to take the patient to surgery if the bleed is in the left deep gray matter)
- Patients with lobar ICH
- Patients with amyloid ICH, on average, do not require surgery.
The goal of surgery is volume reduction to less than 20 cc.
Finally, the question of elevated blood pressure in aneurysmal subarachnoid hemorrhage (SAH) must be addressed. There is concern that the patient may rebleed if blood pressure is high and/or pulse pressure is large. However, lower GCS patients may require higher perfusion pressures to prevent infarction.
For hypertension treatment in acute SAH:
- If the patient has a GCS score of 3 to 6 (World Federation of Neurological Surgeons scale18a grade 5) or has a history of hypertension, maintain SBP <140 mm Hg.
- If the patient has a GCS score >6 and no history of hypertension, maintain SBP <120 mm Hg.
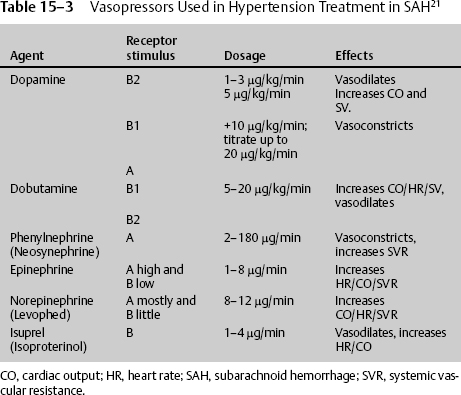
At times the patient’s blood pressure or CPP is too low despite adequate fluid resuscitation. There is no consensus regarding which vasopressor to use. Small-dose IV vasopressin infusion may be beneficial in patients with acute brain injuries and with unstable hemodynamics who are refractory to fluid resuscitation and catecholamine vasopressors.19 Biestro et al20 commented that noradrenaline at a dose of 0.5 to 5.0 mg/hour is effective and safe and might be considered the drug of choice, whereas dopamine was not as effective at a high dose of 10.0 to 42.5 μg/kg/minute, and methoxamine given as a bolus controls sudden decreases in MAP. The use of vasoconstrictor drugs to increase CPP may impair oxygenation in the ischemic penumbra and to other tissues of the body, such as the intestinal mucosa and the kidneys. Several human and animal studies suggest that norepinephrine improves CBF better than dopamine at high doses (Table 15–3).21
 Cerebral Oxygenation
Cerebral Oxygenation
The brain cells need oxygen and need adenosine triphosphate (ATP). In an injured state, the brain does not respond well to blood pressure or volume that is excessively elevated. Blood pressure that is too high will cause cerebral edema, bleeding, and increased ICP. Therefore, a dilemma exists as to the exact titration of therapy; this dilemma has been answered with the introduction of technology that allows the monitoring of cerebral metabolism and brain oxygenation. The literature is replete with documentation that supernormal PaO2 improves brain tissue oxygenation and outcome in hundreds of patients.9,22–35 The hemoglobin molecule has the capacity to carry 17 to 21 mL O2/100 mL blood when all four binding sites are saturated. PaO2 reflects only free oxygen molecules dissolved in plasma and not those bound to hemoglobin. Neither PaO2 nor SaO2 reflects total oxygen in the blood. At 100% saturation, pO2 can be as low as 80 mm Hg; at 75% saturation, pO2 can be as low as 40 mm Hg; and at 50% saturation, pO2 can be as low as 27 mm Hg. It must be remembered that blood cells plus plasma carry more oxygen than can be accounted for by the hemoglobin molecule. The red blood cells (RBCs) reach only 85% of the brain cells, with plasma supplying the remainder. Treating the patient must account for decreased affinity of hemoglobin from increased temperature, pCO2, 2,3-diphospoglycerate, and a decrease in pH. A rightward shift, by definition, causes a decrease in the affinity of hemoglobin for oxygen. This makes it harder for the hemoglobin to bind to oxygen (requiring a higher partial pressure to achieve the same oxygen saturation), but it makes it easier for the hemoglobin to release bound oxygen. Physiologically, increasing FiO2 past the maximal saturation of hemoglobin only increases the dissolved O2 in plasma by 2 to 3%. Furthermore, FiO2 levels higher than 60% may be harmful when used for longer than 24 hours in adults.
Increasing PO2 to higher levels than necessary to saturate hemoglobin drives O2 into the oxygen-starved brain tissue. With supernormal pO2, during the early period after severe head injury, the lactate levels in brain tissue are reduced, and outcome is improved. Brain-injured patients may benefit from 100% O2 during the first 8 to 24 hours, PaO2 >150 mm Hg for 2 to 4 days during peak edema, and >100% thereafter, each in an attempt to maintain brain tissue oxygenation around 40 mm Hg and not below 20 mm Hg. Cerebral tissue monitoring systems measure four areas of brain metabolism, as well as physiological information about the progression of secondary injury at the cellular level, which may indicate the onset of ischemic injury. Experiments have demonstrated good recovery with mean brain partial oxygen pressure at 39±4 mm Hg, moderate to severe disability with mean brain partial oxygen pressure at 31±5 mm Hg, and dead or vegetative disability with a mean brain partial oxygen pressure at 19±8 mm Hg.23–35
Cerebral perfusion must be provided in a tightly regulated manner. The blood pressure must be maintained so that appropriate CPP and CBF continue without causing additional edema, increased ICP, or bleeding. Oxygen has to be supplied in supernormal levels to prevent the ischemic penumbra from infarcting.
< div class='tao-gold-member'>







