 The heat shock response and heat shock proteins provide protection against a broad variety of cellular stresses and injuries.
The heat shock response and heat shock proteins provide protection against a broad variety of cellular stresses and injuries. The heat shock response and heat shock proteins are potent modulators of inflammation-associated signal transduction.
The heat shock response and heat shock proteins are potent modulators of inflammation-associated signal transduction. Heme oxygenase plays a broad range of cytoprotective roles, and a large portion of this protective effect seems to be mediated by carbon monoxide, which is a primary end product of heme oxygenase-mediated degradation of heme.
Heme oxygenase plays a broad range of cytoprotective roles, and a large portion of this protective effect seems to be mediated by carbon monoxide, which is a primary end product of heme oxygenase-mediated degradation of heme. Extracellular heat shock proteins have emerged as important signaling molecules in innate immunity and other cellular processes.
Extracellular heat shock proteins have emerged as important signaling molecules in innate immunity and other cellular processes. Ischemic preconditioning describes a specific form of cellular adaptation whereby brief periods of nonlethal ischemia confer cellular resistance to subsequent and otherwise lethal periods of ischemia.
Ischemic preconditioning describes a specific form of cellular adaptation whereby brief periods of nonlethal ischemia confer cellular resistance to subsequent and otherwise lethal periods of ischemia. Hypoxic preconditioning is a similar concept to that of ischemic preconditioning, but it is specific to hypoxic conditions, rather than ischemia.
Hypoxic preconditioning is a similar concept to that of ischemic preconditioning, but it is specific to hypoxic conditions, rather than ischemia. Hypoxia inducible factor (HIF) is a key molecule in cellular adaptations to hypoxia.
Hypoxia inducible factor (HIF) is a key molecule in cellular adaptations to hypoxia. Endotoxin tolerance describes an intriguing adaptation whereby cellular exposure to low levels of endotoxin reprograms the cellular response to subsequent exposure to higher levels of endotoxin.
Endotoxin tolerance describes an intriguing adaptation whereby cellular exposure to low levels of endotoxin reprograms the cellular response to subsequent exposure to higher levels of endotoxin. The clinical significance of endotoxin tolerance to clinical medicine and innate immunity remains unclear at the present time.
The clinical significance of endotoxin tolerance to clinical medicine and innate immunity remains unclear at the present time. Reactive oxygen species (ROS) are inevitably derived from the requisite use of oxygen by aerobic organisms, and excessive amounts of ROS can lead to oxidative cellular and tissue injury.
Reactive oxygen species (ROS) are inevitably derived from the requisite use of oxygen by aerobic organisms, and excessive amounts of ROS can lead to oxidative cellular and tissue injury. The major antioxidant mechanisms that serve to protect against these ROS include superoxide dismutase, catalase, glutathione peroxidase, thioredoxin, and thioredoxin reductase.
The major antioxidant mechanisms that serve to protect against these ROS include superoxide dismutase, catalase, glutathione peroxidase, thioredoxin, and thioredoxin reductase. Epigenetics refers to heritable changes in gene expression patterns that are not related to direct changes to the DNA sequence of a given gene.
Epigenetics refers to heritable changes in gene expression patterns that are not related to direct changes to the DNA sequence of a given gene. Epigenetic modifications are increasingly being described for immunity- and inflammation-related genes and may have important functional consequences for recovery from critical illness.
Epigenetic modifications are increasingly being described for immunity- and inflammation-related genes and may have important functional consequences for recovery from critical illness. variety of biologic stresses (1,2,3). The heat shock response is
variety of biologic stresses (1,2,3). The heat shock response is characterized by the rapid expression of a unique group of proteins known collectively as heat shock proteins (HSPs), when a cell or organism is exposed to environmental stress. The structure, mode of regulation, and function of HSPs are highly conserved among different species, and HSPs have been identified in virtually all eukaryotic and prokaryotic species examined. While classically described as a response to heat stress (hence the term heat shock response), HSPs can be induced by a wide variety of nonthermal stressors and pharmacologic agents (Table 20.1). For this reason, the terms “stress response” and “stress proteins” may be descriptors that are more appropriate. Many of these stimuli are relevant to the critically ill patient, and it is now well established that critically ill patients readily express HSPs (i.e., mounting a heat shock response) (4,5,6,7). Moreover, and, particularly germane to pediatric critical care medicine, the experimental literature suggests that younger animals have a greater capacity to express HSPs than older animals (8). Indeed, the loss of the ability to generate a stress response may contribute to the aging process (9,10).
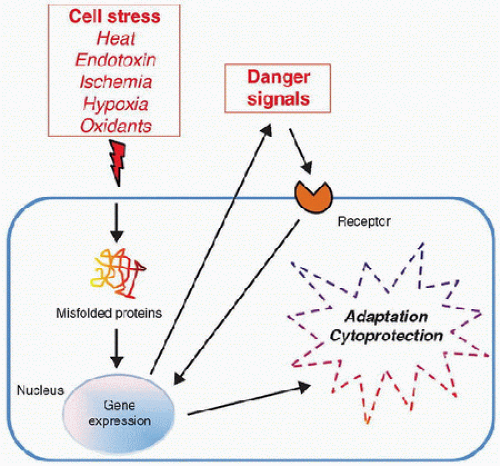 FIGURE 20.1. Simplified representation of the basic mechanisms of cellular adaptation. Exposure to stress (e.g., heat, endotoxin, ischemia, hypoxia, oxygen radicals, etc.) results in perturbations of the intracellular milieu (e.g., stress-induced misfolding of intracellular proteins) that trigger new gene expression and/or the release of danger signals that activate stress responses via receptor-mediated signal transduction. In either case, the reprograming of gene expression allows for cellular adaptations to stress and cytoprotection. |
(discussed in detail later in this chapter). In addition, ubiquitin, inhibitor of κB (IκB), endothelial nitric oxide synthase (eNOS), and mitogen-activated protein kinase phosphatase-1 (MKP-1) are listed as HSPs in that they can be induced by classic heat shock. Many HSPs are constitutively expressed (e.g., HSP90 is one of the most abundant proteins found inside the cell), while others are rapidly and highly expressed in response to cellular stress. Among the latter, HSP72 is one of the more well-studied inducible HSPs in the context of cellular adaptations to stress and consequent protection against cellular injury (see in what follows).
TABLE 20.1 INDUCERS OF THE STRESS RESPONSE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 20.2 THE MAJOR FAMILIES OF HEAT SHOCK PROTEINS | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 transduction pathway (14). NF-κB is a transcription factor that regulates the expression of many genes involved in
transduction pathway (14). NF-κB is a transcription factor that regulates the expression of many genes involved in inflammation (see Chapter 19). Heat shock-mediated inhibition of the NF-κB pathway involves inhibition of IκB kinase and increased de novo expression of the endogenous NF-κB inhibitory protein, IκBα. These inhibitory effects of heat shock on cellular proinflammatory responses and the NF-κB pathway appear to be relatively specific, rather than a global downregulation of cellular function and gene expression. Furthermore, genome-level studies indicate that the mononuclear cell response to heat stress is highly divergent compared with the mononuclear cell response to endotoxin (15).
 cytoprotection (16,18). For example, overexpression of HO-1 conferred protection against oxygen toxicity, hemoglobin toxicity, tumor necrosis factor (TNF)-α-mediated apoptosis, and Pseudomonas-mediated cellular injury and apoptosis. Experiments in animal models, involving either pharmacologic induction of HO-1 or genetic overexpression of HO-1, confirm that HO-1 confers cytoprotection in vivo. Induction of HO-1, by intravenous hemoglobin administration, protected rats against the lethal effects of endotoxemia. Lung epithelial overexpression of HO-1, via an adenovirus vector, conferred protection in rats exposed to hyperoxia; cardiac-specific overexpression conferred protection in a murine model of ischemia. There is also interest in HO-1-mediated cytoprotection in the field of transplant biology. In a cardiac xenograft transplantation model, increased expression of HO-1 improved graft survival.
cytoprotection (16,18). For example, overexpression of HO-1 conferred protection against oxygen toxicity, hemoglobin toxicity, tumor necrosis factor (TNF)-α-mediated apoptosis, and Pseudomonas-mediated cellular injury and apoptosis. Experiments in animal models, involving either pharmacologic induction of HO-1 or genetic overexpression of HO-1, confirm that HO-1 confers cytoprotection in vivo. Induction of HO-1, by intravenous hemoglobin administration, protected rats against the lethal effects of endotoxemia. Lung epithelial overexpression of HO-1, via an adenovirus vector, conferred protection in rats exposed to hyperoxia; cardiac-specific overexpression conferred protection in a murine model of ischemia. There is also interest in HO-1-mediated cytoprotection in the field of transplant biology. In a cardiac xenograft transplantation model, increased expression of HO-1 improved graft survival.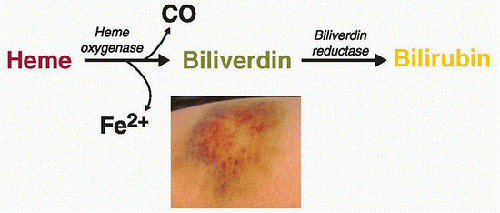 FIGURE 20.2. HO-mediated degradation of heme. HO degrades heme to biliverdin and concomitantly generates CO and iron (Fe). Biliverdin is subsequently converted to bilirubin via biliverdin reductase. The respective colorimetric reactions that coincide with the generation of these heme degradation products are readily evident in the various hues of a common bruise. |
For example, adult patients suffering from major trauma have increased serum levels of HSP72 (4). Critically ill children with septic shock also have increased serum levels of HSP72, and the absolute levels are much higher than that reported for critically ill adult patients following multisystem trauma (5). The latter may be reflective of an increased capacity of children to express HSPs compared with adults, consistent with the experimental literature previously mentioned.
 “danger signals” for the innate immune system.
“danger signals” for the innate immune system.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


