KEY POINTS
1. The heart has a fibrous skeleton that provides an insertion site at each valvular ring. This fibrous structure also connects cardiac myocytes so that “stretch” or preload results in sarcomeres lengthening and not intercellular sliding.
2. Contractility changes as a result of myoplasmic Ca2+ concentration change during systole.
3. Relaxation following contraction is an active process requiring ATP consumption to pump Ca2+ into the sarcoplasmic reticulum as well across the sarcolemma.
4. The fundamental unit of tension development is the sarcomere.
5. The endocardium receives perfusion during systole while the epicardium receives blood flow throughout the cardiac cycle. The ventricular wall is more susceptible to endocardial infarction.
6. The external stroke volume work the ventricle does is to raise the pressure of a stroke volume from VEDP (right or left) to mean arterial pressure (pulmonary or systemic, respectively).
7. Oxygen or ATP consumption occurs during release of actin–myosin bonds during relaxation of this bond. The main determinants of myocardial oxygen consumption are heart rate, contractility, and wall tension.
8. The cardiovascular system regulates blood pressure and it is an example of a negative feedback loop control system. Sensors throughout the cardiovascular system all sense stretch.
9. Physiologic reserves are expansion factors allowing the cardiovascular system to maintain blood pressure. These reserves are heart rate (3-fold range), contractility, systemic vascular resistance (15-fold range), and venous capacitance (~1.5-fold range).
10. Utilizing the reserve of venous capacitance does not affect myocardial oxygen consumption.

AS A PHYSIOLOGIC PRIMER FOR CARDIAC ANESTHESIOLOGY, this chapter requires brevity and choices (or opinions)! Cardiac anatomy, physiology, pathology, and genomics are decades old, continue to evolve, and have a vast literature. Our focus is on presenting physiologic principles important to clinical management in the operating room. A detailed description and discussion of cardiac physiology can be found in 1. If these physiologic concepts are memorized and applied correctly, your patients will benefit from risk modification under your care. If they are understood, you will be able to apply them to the dynamic pathology present in the operating room and have a physiologic basis for being a consultant in cardiac anesthesiology.
I. Embryologic development of the heart.
A. The cardiovascular system begins to develop during week 3 as the primitive vascular system is formed from mesodermally derived endothelial tubes. Eventually at week 4, bilateral cardiogenic cords from paired endocardial heart tubes fuse into a single heart tube (primitive heart). This fusion initiates forward flow and is the start of the heart’s transport function.
B. The primitive heart evolves into four chambers: Bulbus cordis, ventricle, primordial atrium, and sinus venosus, eventually forming bulboventricular loop with the initial contraction occurs at 21 to 22 days. These contractions result in unidirectional blood flow in week 4.
C. From weeks 4 to 7 heart development enters a critical period as it divides into the four chambers of the adult heart, the basis of the fetal circulation.
D. The framework of the heart is a fibrous skeleton, composed of fibrin and elastin, forming four rings encircling the four valves of the heart as well as intermyocyte connections.
E. The fibrous skeleton:
1
1. Serves as an anchor for the insertion of the valve cusps
2. Resists overdistention of the annuli of the valves (resisting incompetence)
3. Provides a fixed insertion point for the muscular bundles of the ventricles
4. Minimizes intermyocyte sliding during ventricular filling and contraction
II. Electrical conduction
A. By breaking the atrial intermyocyte conduction of impulses, the fibrous skeleton blocks the direct spread of electrical conduction from the atria to the ventricles.
B. Because of this separation of the atrial myocyte conduction from ventricular myocytes, the AV nodal/purkinje system creates coordinated contraction between atria and ventricles.
C. Excitation–contraction coupling
1. A purkinje fiber action potential results in the coordinated contraction of a cardiac myocyte. There are five phases of an action potential and an integrated and complex change in Na+, K+, and Ca2+ conductances and ion fluxes within these phases.
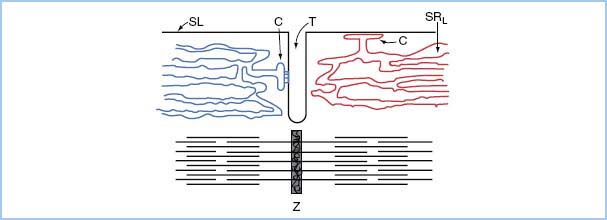
2. Excitation–contraction coupling starts when an action potential triggers the diffusion of Ca2+ ions across the sarcolemma through the Ca2+ release channels (ryanodine receptor channel). To shorten the time constant of this transmembrane flux, the T-tubule system markedly increases the surface area through which this flux of external Ca2+ occurs. In addition, the sarcoplasmic reticulum cisternae ensure rapid myoplasmic Ca2+ increase (Fig. 1.1).
3. The amount of Ca2+ transported across the sarcolemma is only about 1% of Ca2+ needed for contraction. However, this Ca2+ serves as a trigger for sarcoplasmic reticulum release of Ca2+ to create the Ca2+ concentration needed for tension development.
4. The sarcoplasmic reticulum is a complex organelle responsible for the efficient cycling of calcium concentration during each heartbeat. The external Ca2+ crossing the sarcolemma triggers a graded release of internal Ca2+ from the sarcoplasmic reticulum. From relaxation to contraction, cytosolic Ca2+ concentration varies approximately 100-fold.
Figure 1.1 Relation of cardiac sarcoplasmic reticulum to surface membrane and myofibrils. The SRL and C overlie the myofilaments; they are shown separately for illustrative purposes. SL, sarcolemma; C, cisterna; T, transverse tubule; SRL, longitudinal sarcoplasmic reticulum; Z, Z disc.
2
5. Three very important proteins within the sarcoplasmic reticulum are responsible for controlling calcium flux: The Ca2+ release channel, the sarco-endoplasmic reticulum ATP-ase (SERCA-2), and the regulatory protein of SERCA-2 (phospholamban). Alterations within phospholamban are currently an active area of interest and may play a role in the development of heart failure and other cardiomyopathies (Maclennan, Nature Reviews Molecular Cell Biology 4, 566–577 [July 2003]).
6. The graded release of Ca2+, signaled by the trans-sarcolemmal Ca2+ flux and enhanced due to sarcoplasmic reticulum cisternae, is dependent on the amount of Ca2+ stored in the sarcoplasmic reticulum, the sympathetic tone (sarcolemma Ca2+ conductance), and the external Ca2+. Increased stores lead to increased release, resulting in increased tension development for a given sarcomere length = increased contractility.
7. Any increase in contractility via any drug is a consequence of increased myoplasmic Ca2+ concentration during systole! Ca2+ binds to troponin and results in a conformational change involving tropomyosin. This change allows actin and myosin to interact, resulting in shortening of a sarcomere (Fig. 1.2).
8. Similarly, a decrease in contractility is a result of decreased Ca2+ binding to troponin during systole. Its cause is attributable to decreased myoplasmic Ca2+ during systole or decreased Ca2+ binding to troponin. Myoplasmic Ca2+ is affected by inhalational agents, induction drugs, etc. A common clinical reason for decreased Ca2+–troponin affinity is intracellular acidosis.
3
9. Finally, for contraction to cease, Ca2+ must be removed from the myoplasm. Ca2+ is actively pumped into the sarcoplasmic reticulum as well as pumped across the sarcolemma. Depending on Ca2+ ion conductances in each of the sites, sarcoplasmic Ca2+ may be increased or decreased in the sarcoplasmic reticulum during this removal.
III. Cardiac myocyte
A. Sarcomere
1. The fundamental unit of tension development is the sarcomere. The sarcomere is composed of myosin, the tropomyosin–actin–troponin complex, and a z-disc.
2. Each actin molecule contains a myosin-binding site as well as the site for tropomyosin–troponin chain to bind. Two actin molecule chains are intertwined and form an actin polymer. Combined with a tropomyosin–troponin complex bound within the grooves to the actin polymer chains, this complex is known as a thin filament. The thin filament spans a distance of approximately 2 μm.
3. The z-disc anchors the thin filaments in place in a regular pattern as schematized below. It is a strong meshwork of filaments forming a band that anchors the interdigitated thin filaments.
Figure 1.2 Schematic representation of actin–myosin dependence on Ca2+–troponin binding for tension development to occur. (From Honig C. Modern Cardiovascular Physiology. Boston/Toronto: Little, Brown and Company; 1981.)
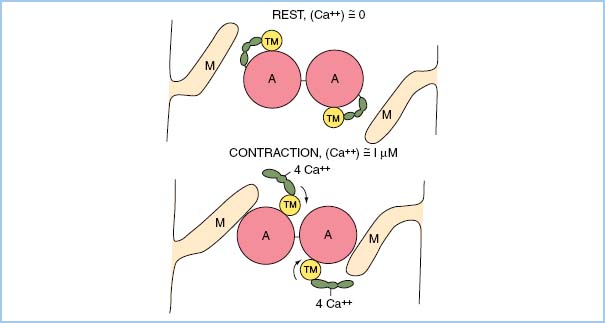
4. Myosin molecules aggregate spontaneously forming the thick filaments. These filaments are approximately 1.6 μm. These thick filaments are held in place by M filaments and are interdigitated amongst the thin filaments. At the center of each thick filament is a zone that has no myosin “heads.” This absence explains the decrease in tension development if overstretching of the sarcomere were to occur. Each myosin head contains an ATP-ase and actin-binding site.
5. Together, troponin and tropomyosin and Ca2+ binding serve as regulatory proteins that allow actin and myosin to interact to result in shortening of the sarcomere length.
6. Each exposure of actin to myosin results in a shortening of sarcomere length. To allow for multiple shortenings to occur, actin–myosin binding is uncoupled. This ATP-consuming step, the relaxation phase, occurs because of the ATP-ase bound to myosin. This energy-dependent step, requiring oxygen consumption, occurs multiple times during a cardiac cycle.
7. Depending on preload, the sarcomere length, or the distance between z-discs, ranges from 1.8 to 2.2 μm.
IV. Organization of myocytes
A. A cardiac myocyte is approximately 12 μm long. Hence, each myocyte only has several (~6) sarcomeres in series from end to end of a myocyte.
B. As mentioned under embryology, collagen fibers link cardiac myocytes together. These collagen fiber links connect adjacent myocytes that hold all myocytes together allowing for a summation of the shortening of each myocyte into a concerted shortening of the ventricle.
C. This collagen structure also limits the cardiac myocyte from being overstretched, minimizing the risk of destroying a cell or limiting actin–myosin exposure through overstretching [1, p. 9].
D. Additional complexity is that from epicardium to endocardium, longitudinal alignment of the cardiac myocytes occurs in layers. Hence, shortening in each layer results in distortion between the layers.
5
E. This distortion results in partial occlusion of penetrating arteries arising from the surface vessels supplying blood to the inner layers of the myocardium.
F. Consequently, the endocardium is more vulnerable to ischemia than the epicardium because blood flow to the endocardium occurs primarily during systole while that to the epicardium occurs during the entire cardiac cycle.
V. Length–tension relationship
A. Consider this thought experiment. There is an idealized, single cardiac myocyte that is 12 μm long with 6 sarcomeres in series. All distances between Z-discs (sarcomere lengths) are equal for each of the 6 sarcomeres (12 μm = 6 * 2 μm). As the cardiac myocyte length changes, the length of each sarcomere changes proportionately. This single myocyte is suspended so that a strain gauge measures the tension that the myocyte generates at rest and during contraction.
1. The idealized myocyte is stretched between two fixed points.
2. The tension caused by the force of stretching the muscle at rest and created by the muscle during contraction is measured.
3. The muscle is stretched at rest over a range of 10.8 to 16.2 μm. Consequently, the sarcomere lengths vary between 1.8 and 2.7 μm as this myocyte contracts.
4. At each sarcomere length, two fixed myoplasmic Ca2+ concentrations are set within the cell: Zero concentration (rest) and a known value (contraction).
5. Myocyte tension is measured for both Ca2+ concentrations.
6. Plotting resting tension as a function of length results in a resting length–tension plot.
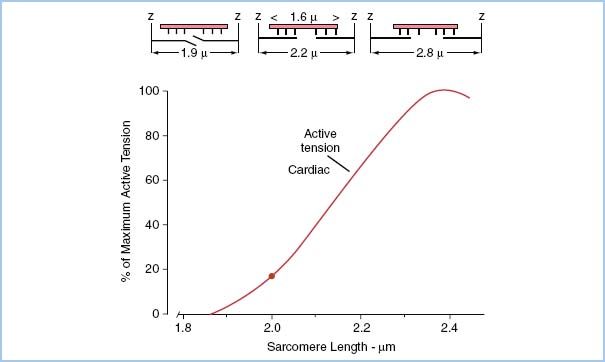
7. Recording peak tension as a function of myocyte length, a length–tension curve can be plotted for the given Ca2+ concentration as depicted in Figure 1.3.
B. Compliance
1. In our idealized myocyte model, a measured amount of tension was required to stretch the sarcomere to a value between 1.9 and 2.6 μm. If we were to plot this passive tension resulting from passive stretch of the sarcomeres, there would be very little passive tension required to stretch the sarcomeres until around 2.2 μm. As the sarcomeres become stretched beyond 2.2 μm in intact cardiac muscle, the fibrous skeleton restricts further stretching resulting in a rapid change in pressure with very little change in volume (very low compliance).
2. This resting relationship between length and tension is the equivalent of ventricular compliance as will be discussed below.
Figure 1.3 Top three schematic diagrams represent actin (thick filaments penetrating Z disc) and myosin (thin filaments forming sheaf between Z disc) filaments at three sarcomere lengths 1.9, 2.2, and 2.8 μm. Bottom graph represents per cent of maximum tension development versus sarcomere length for strips of cardiac muscle. Note that fibrous cardiac skeleton inhibits sarcomere stretch from approaching a sarcomere length of 2.8 μm. (Modified from Honig C. Modern Cardiovascular Physiology. Boston/Toronto: Little, Brown and Company; 1981.)
C. Contractility
1. For a given Ca2+ concentration, a fraction of troponin molecules will bind Ca2+ molecules.
2. This Ca2+ binding to each troponin results in a conformational change in its corresponding tropomyosin that allows an actin and myosin head pair, opposing each other and regulated by these tropomyosin molecules, to interact.
3. In addition to this conformational change, the percentage of the actin and myocytes heads opposed to each other is dependent on sarcomere length.
4. If one repeats the above mental experiment with a different known Ca2+ concentration, a new contracting length–tension curve is plotted.
5. For a range of Ca2+ concentrations in cardiac myocytes, a family of length–tension curves results.
6. Only through a change in myoplasmic Ca2+ concentration during contraction can contractility change.
D. Intracellular Ca2+ concentration
1. The range of myoplasmic Ca2+ concentrations during contraction varies depending upon Ca2+ fluxes across the sarcolemma at the initiation of contraction and the Ca2+ released from the sarcoplasmic reticulum.
2. The flux of Ca2+ across the sarcolemma is only ~1% of Ca2+ present during contraction. However, changes in this 1% result in changes in the amount of Ca2+ released from the sarcoplasmic reticulum.
3. The sarcoplasmic reticulum response is graded. The more the Ca2+ crossing the sarcolemma, the more the Ca2+ released from the sarcoplasmic reticulum.
4. Examples of increasing sarcolemmal Ca2+ flux include increasing epinephrine levels and increasing external Ca2+ concentration (a CaCl2 bolus).
5. Increased sarcoplasmic reticulum Ca2+ stores result from increased Ca2+ flux across the sarcolemma and increased heart rate (HR).
E. Oxygen consumption
1. Each interaction of actin and myosin results in a submicron shortening of the sarcomere. For the sarcomere to shorten 15%, many actin–myosin interactions take place.
2. Therefore, shortening requires a repetitive interaction of actin–myosin complexes.
3. Each interaction requires ATP for release of the actin–myosin head. It is relaxation of the actin–myosin interaction that requires energy and consumes oxygen.
4. Remember that the more actin–myosin cycles in a unit of time, the more the oxygen consumption!
VI. A heart chamber and external work
A. The chamber wall
1. To form a ventricular chamber, individual myocytes are joined together via collagen fibrin strands. This joining of myocytes, along a particular direction but not end to end, results in a sheet of muscle with myocytes oriented along a similar axis.
2. Several such layers form the ventricular wall. These layers insert on the valvular annuli.
3. Because of the electrical distribution of the signal through the purkinje system, the layers contract synchronously resulting in shortening of the muscle layers and a reduction in the volume of the chamber itself.
B. Atria
1. Atrial contraction contributes approximately 20% of the ventricular filling volume in a normal heart and may contribute even more when left-ventricular end-diastolic pressure (LVEDP) is increased. In addition to the volume itself, the rapid rate of ventricular volume addition resulting from atrial contraction may play a role in ventricular sarcomere lengthening.
C. Ventricle
1. For a given state of contractility (myoplasmic Ca2+ concentration), sarcomere length determines the wall tension the ventricle can achieve as discussed above. The aggregate shortening of the sarcomeres in the layers of cardiac myocytes results in wall tension that leads to ejection of blood into the aorta and pulmonary artery (PA).
2. The active range of sarcomere length is only 1.9 to 2.2 μm or ~15% of its length. Falling below 1.8 μm results from an empty ventricle and an empty heart cannot pump blood. The collagen fiber network inhibits the stretching of sarcomeres much above 2.2 μm. This integration of structure and function is important in permitting survival. If there were no skeleton, overstretch would lead to reduced emptying that would lead to more overstretch and no cardiac output (CO).
D. Preload and compliance
1. Preload
a. Where clinicians speak of preload, muscle physiologists think of sarcomere length. Clinician’s surrogate for initial sarcomere length is end-diastolic ventricular volume, and not ventricular pressure.
b. As discussed above, the sarcomere length determines how many actin–myosin heads interact for a given myoplasmic Ca2+ concentration at any instant.
c. Measuring sarcomere length is essentially impossible clinically. As a surrogate indirect estimate of sarcomere length, clinicians measure a chamber pressure during chamber diastole. This pressure measurement is the equivalent of the myocyte tension measurement above.
d. A more direct surrogate estimate of sarcomere length is chamber volume. As echocardiography is commonly available, estimates of volumes are direct estimates of preload and remove assumptions about chamber compliance that are necessary from the estimate when chamber pressure is used.
e. A plot of the relationship between chamber pressure and chamber volume results in a curve similar to the resting length tension curve for the myocyte.
f. The slope of this curve at any point (change in volume over the change in pressure at that point) is the compliance of the chamber at that point. The pressure–volume curve is nonlinear, and this slope varies depending on ventricular volume. Ventricles become much less compliant as sarcomere length surpasses 2.2 μm because of the collagen fiber skeleton.
g. Non-ischemic changes in compliance generally occur over long time periods. However, ischemia can change ventricular compliance very quickly. Thick ventricles, ventricles with scar formation, or ischemic ventricles have a lower compliance than normal ventricles. Less compliant ventricles require a higher pressure within them to have equal volume within them compared to a more compliant ventricle.
h. While preload is most commonly considered in the left ventricle, it is important in all four chambers. Congenital heart disease and cardiac tamponade can make that very apparent.
E. Ventricular work
1. For a sarcomere length between 1.9 and ~2.2 μm (preload) with a given systolic myoplasmic Ca2+ concentration (contractility), the cardiac myocyte will shorten, develop tension that increases with increased sarcomere length, and eject blood from the ventricle—a stroke volume (SV).
2. In ejecting this SV the ventricle performs external work. This external work is the raising of SV from LVEDP to the ventricular pressure in systole. In the absence of valvular heart disease, the systolic pressure and ventricular pressure in systole are closely matched by those in the aorta or PA for the respective ventricles.
3. Normal blood pressures (BPs) in the aorta and PA are not dependent on subject size and vary little amongst normal subjects. Normalizing SV to stroke volume index (SVI), SV divided by body surface area (BSA), the variability amongst subjects is small.
6
4. Formally, the external work of a ventricle is the area within the pressure volume loop seen in Figure 1.4. The definition of various points and intervals is defined in the legend. We estimate this indexed work for the left ventricle (LVSWI) by multiplying the SVI in milliliter times the difference in arterial mean pressure and ventricular pressure at the end of diastole (commonly estimated as atrial pressure or PA wedge pressure) times a constant (0.0136) to convert to clinical units:
LVSWI = SVI * (SBP mean − LVEDP) * 0.0136 (g m/m2)
Figure 1.4 Idealized pressure–volume loop. Area within the loop represents LVSW. Dividing SV (Point B minus Point A volumes) by BSA results in SVI. The area within this indexed loop is the LVSWI.

5. The normal resting values for SVI and LVSWI are ~50 mL/m2 and 50 g m/m2.
6. In performing this work, the efficiency of the ventricle, the ratio of external work done to energy consumed to do it, approaches that of a gasoline engine—only 10%. An astonishingly inefficient process given that our lives depend on it (and we cannot improve it)!
7. Since external work includes the product of pressure difference and SV, the amount of work does not distinguish between these two variables. Evidence suggests that the ventricles can do volume work somewhat more efficiently (require less oxygen) than pressure work. The reasoning relates to how many actin–myosin cycles are required to shorten a sarcomere to a given distance! The hypothesis is that it takes fewer actin–myosin cycles to shorten the same distance for volume work compared to pressure work. This principle may explain an underlying reason for success using vasodilators to treat heart failure.
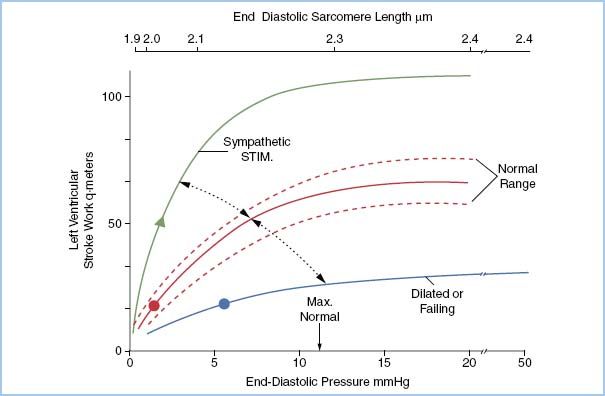
F. Starling curve
1. For a given myoplasmic Ca2+ concentration (contractile state), varying the sarcomere length between 1.9 μm and less than 2.2 μm increases the amount of external work changes. For a ventricle with a normal compliance, a sarcomere length of 2.2 μm corresponds to one with an LVEDP of 10 mm Hg.
7
2. By plotting the relationship of ventricular pressure with left ventricular work index, a Starling curve is generated.
3. By changing the contractile state and replotting the same relationship, a new curve develops resulting in a family of Starling curves idealized in Figure 1.5.
G. Myocardial oxygen consumption
1. Except for very unusual circumstances, substrate for ATP and phosphocreatine (PCr) production is readily available. At any given moment the intracellular oxygen content is capable of keeping the heart contracting for seconds but the carbohydrate and lipid store can fuel the heart for almost an hour. Hence, capillary blood flow is crucial to maintain oxidative metabolism.
2. Myoglobin is an intracellular oxygen store. Its affinity for oxygen is between hemoglobin and cytochrome aa3. The maximum oxygen concentration required in mitochondria for maximal ATP production is 0.1 Torr! Myoglobin concentration is high enough to buffer interruptions in capillary flow only for seconds. Compared to high-energy phosphate buffers, this time buffer is small relative to ATP consumption rates. However, its intermediate oxygen affinity enhances unloading from the red cell into the myocyte and also serves to distribute oxygen within the cell.
3. Commonly, blood flow to regions of the heart limits oxygen delivery resulting in decreased. ATP production will result in reduced wall motion in that region—regional ischemic heart disease.
Figure 1.5 The Starling curve.
Related posts:
 Devices for Cardiac Support and Replacement
Devices for Cardiac Support and Replacement
 Alternative Approaches to Cardiac Surgery with and without Cardiopulmonary Bypass
Alternative Approaches to Cardiac Surgery with and without Cardiopulmonary Bypass
 Anesthetic Considerations for Patients with Pericardial Disease
Anesthetic Considerations for Patients with Pericardial Disease
 Anesthetic Management for Patients with Congenital Heart Disease: The Pediatric Population
Anesthetic Management for Patients with Congenital Heart Disease: The Pediatric Population
 Devices for Cardiac Support and Replacement
Devices for Cardiac Support and Replacement
 Anesthetic Management for Patients with Congenital Heart Disease: The Adult Population
Anesthetic Management for Patients with Congenital Heart Disease: The Adult Population
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


