KEY POINTS
1. Drug errors are a common cause of accidental injury to patients. The author suggests referring to drug package inserts or the Physician Desk Reference to check any unfamiliar drug before it is prescribed or administered.
2. Phenylephrine can be used for hypotension related to low systemic vascular resistance states or as a temporizing therapy for hypovolemia.
3. Epinephrine is a direct α1 and α2 and b1 and b2 agonist not dependent on release of endogenous norepinephrine. In the setting of a dilated left ventricle and myocardial ischemia, epinephrine may increase coronary perfusion pressure and reduce ischemia.
4. Milrinone increases intracellular concentration of cAMP. Milrinone used as a single inotropic agent has favorable effects on myocardial supply/demand balance reducing preload and afterload and has a low tendency to produce tachycardia.
5. Nitroglycerine is a direct vasodilator producing greater venous pooling than arterial dilation. Venous pooling caused by dilation decreases heart size and preload reducing MV02 and usually lessens ongoing ischemia.

NUMEROUS POTENT DRUGS ARE USED to control heart rate (HR) and rhythm, blood pressure (BP), and cardiac output before, during, and after surgery. Patients undergoing cardiovascular and thoracic operations are particularly likely to receive one of these agents. This chapter reviews the indications, mechanisms, dosing, drug interactions, and common adverse events for these drugs. Drug errors are common causes of accidental injury to patients, particularly in hospitalized, critically ill patients. Therefore, we suggest that the package insert or Physicians’ Desk Reference [1] (which contains the package insert information) be consulted before any unfamiliar drug is prescribed or administered [2]. Fortunately, it has never been easier to obtain drug information. Convenient sources of drug information include numerous books and web sites, some of which are provided at the end of this chapter [2–5]. Using a “smart” mobile telephone or tablet computer, many physicians now maintain a readily updated library of drug information in their hand or pocket at all times.
1
I. Drug dosage calculations
A. Conversions to milligram or microgram per milliliter
1. Drugs are administered in increments of weight or units. Unfortunately, drugs are not labeled in a uniform manner. Dilution of drugs and calculations are often necessary. Fortunately, most modern infusion pumps do these calculations for the operator, limiting the opportunity for mistakes.
2. A drug labeled z% contains z g/dL; 10 × z equals the number of grams per liter, numerically equivalent to the number of milligrams per milliliter.
a. Example: Mannitol 25% solution contains 25 g/dL, which equals 250 g/L or 250 mg/mL.
b. Example: Lidocaine 2% contains 2 g/dL, or 20 g/L, or 20 mg/mL.
3. Concentrations given as ratios are converted to milligrams or micrograms per milliliter as follows:
1:1,000 = 1 g/1,000 mL = 1 mg/mL
1:1,000,000 = 1 g/1 million mL = 1 μg/mL
a. Example: Epinephrine is packaged for resuscitation in 1:10,000 dilution. Thus, it is one-tenth as concentrated as 1:1,000; therefore, 1:10,000 is 0.1 mg/mL (or 100 μg/mL).
b. Example: A brachial plexus block is to be performed with 0.5% bupivacaine to which epinephrine 1:200,000 must be added. Because the desired concentration is five times greater than 1:1,000,000, 5 μg of epinephrine should be added for each milliliter of bupivacaine.
B. Calculating infusion rates using standard drip concentrations (adults)
1. Step 1. Dose rate (μg/min): Calculate the desired per-minute dose. Example: A 70 kg patient who is to receive dopamine at 5 μg/kg/min needs a 350 μg/min dose rate.
2. Step 2. Concentration (μg/mL): Calculate how many micrograms of drug are in each milliliter of solution. To calculate concentration (μg/mL), simply multiply the number of milligrams in 250 mL by 4. Example: When nitroglycerin is diluted 100 mg/250 mL, there are 100 × 4 = 400 μg/mL. Example: Dopamine, 200 mg, added to 250 mL fluid = 200/250 mg/mL = 800 mg/L = 800 μg/mL concentration.
3. Step 3. Volume infusion rate (mL/min): Divide the dose rate by the concentration (μg/min ÷ μg/mL = mL/min). The infusion pump should be set for this volume infusion rate. Example: 350 μg/min ÷ 800 μg/mL = 0.44 mL/min. Conversion of volume rate from milliliters per minute to milliliters per hour simply involves multiplying by 60 min/h (0.44 mL/min × 60 = 26 mL/h).
4. Table 2.1 can be consulted as an alternative means for determining vasoactive drug infusion rates for patients of different weights.
5. Finally, and perhaps most importantly, the wide availability of micropressor-controlled, programmable infusion pumps has largely eliminated the need for complex calculations at the bedside.
C. Preparation of drug infusions for pediatric patients
1. Most pediatric anesthesia departments, critical care units, and pharmacies have specific preferences as to how drugs should be mixed prior to infusion. We strongly recommend that practitioners adhere to the predominant practice in their unit, whether or not the practitioner may perceive it to be optimal. In general, patient safety is maximized when variation is minimized. We discuss two of the more common ways in which drugs are diluted for pediatric patients in the succeeding sections.
2. Technique A: Standard, single drug concentration for all patients
a. Advantages are that it is simple (no arithmetic calculations are required) and the fluid volume administered scales upward appropriately with weight.
b. Disadvantages are that the standard dilution for each drug must be remembered, and volume infusion rates may be excessive in critically ill infants.
3. Technique B: Custom drug dilution based on patient weight. This method permits infusion of a single fluid volume rate to patients of any weight. Our opinion is that this technique maximizes the possibility for drug dilution mistakes.
Table 2.1 Vasoactive drug infusion rates
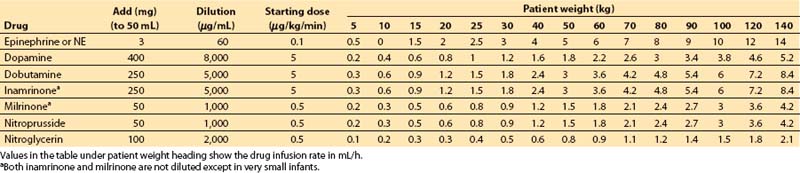
a. Step 1. Decide on a starting dose per kilogram for the drug. Some standard values are as follows:

b. Step 2. Multiply starting dose (in μg/kg/min) by weight (in kg) to give starting dose rate in μg/min.
c. Step 3. Decide on volume rate of fluid that should carry this starting dose of drug into the patient:

These volumes may be decreased substantially if a continuous carrier infusion is utilized.
d. Step 4. Divide starting dose rate (step 2) by volume rate (step 3) to give desired concentration of drug. Units cancel: (μg/min)/(mL/min) = μg/mL.
Example: In a 6.3 kg baby
(1) Select standard starting dosages of dopamine and isoproterenol.
(2) Calculate starting dose rate:
(a) Dopamine: 5 μg/kg/min × 6 kg = 30 μg/min.
(b) Isoproterenol: 0.1 μg/kg/min × 6 kg = 0.6 μg/min.
(3) Choose volume rate: 0.05 mL/min.
(4) Calculate concentration:
(a) Dopamine: 30 μg/min ÷ 0.05 mL/min = 600 μg/mL.
(b) Isoproterenol: 0.6 μg/min ÷ 0.05 mL/min = 12 μg/mL.
(5) Dilute drugs:
(a) Dopamine: 600 mg/L (or 150 mg/250 mL).
(b) Isoproterenol: 12 mg/L (or 3 mg/250 mL).
D. Pediatric resuscitation doses. We find it convenient to prepare syringes of certain drugs (e.g., epinephrine and atropine) so that they contain a standard emergency dose for the patient.
II. Drug receptor interactions
A. Receptor activation. Can responses to a given drug dose be predicted? The short answer is: Partially. The more accurate answer is: Not with complete certainty. Many factors determine the magnitude of response produced by a given drug at a given dose.
1. Pharmacokinetics relates the dose to the concentrations that are achieved in plasma or at the effect site. In brief, these concentrations are affected by the drug’s volume of distribution and clearance, and for drugs administered orally, by the fractional absorption [6,7].
2. Pharmacodynamics relates drug concentrations in plasma or at the effect site to the drug effect.
a. Concentration of drug at the effect site (receptor) is influenced by the concentration of drug in plasma, tissue perfusion, lipid solubility, and protein binding; diffusion characteristics, including state of ionization (electrical charge); and local metabolism.
b. Number of receptors per gram of end-organ tissue varies.
(1) Upregulation (increased density of receptors) is seen with a chronic decrease in receptor stimulation. Example: Chronic administration of β-adrenergic receptor antagonists increases the number of β-adrenergic receptors.
(2) Downregulation (decreased density of receptors) is caused by a chronic increase in receptor stimulation. Example: Chronic treatment of asthma with β-adrenergic receptor agonists reduces the number of β-adrenergic receptors.
c. Drug receptor affinity and efficacy may vary.
(1) Receptor binding and activation by an agonist produces a biochemical change in the cell. Example: α-Adrenergic receptor agonists increase protein kinase C concentrations within smooth-muscle cells. β-Adrenergic receptor agonists increase intracellular concentrations of cAMP.
(2) The biochemical change may produce a cellular response. Example: Increased intracellular protein kinase C produces an increase in intracellular [Ca2+], which results in smooth-muscle contraction. Conversely, increased intracellular concentrations of cAMP relax vascular smooth muscle but increase the inotropic state of cardiac muscle.
(3) The maximal effect of a partial agonist is less than the maximal effect of a full agonist.
(4) Receptor desensitization may occur when prolonged agonist exposure to receptor leads to loss of cellular responses with agonist–receptor binding. An example of this is the reduced response to β1-adrenergic receptor agonists that occurs in patients with chronic heart failure (CHF), as a result of the increased intracellular concentrations of β-adrenergic receptor kinase, an enzyme which uncouples the receptor from its effector enzyme adenylyl cyclase.
(5) Other factors including acidosis, hypoxia, and drug interactions can reduce cellular response to receptor activation.
III. Pharmacogenetics and genomics
In the future, pharmacogenetics, or how drug actions or toxicities are influenced by an individual’s genetic make-up, may become a tool in helping anesthesiologists select among therapeutic options. We are learning, for example, that the genetic profile of the individual may impact the degree to which patients respond to adrenergic therapies, including vasopressors. Life-threatening arrhythmias, such as Long QT syndrome, may result from therapy with a number of commonly prescribed agents, and relatively common genetic sequence variations are now known to be an underlying predisposing factor. Droperidol, which is highly effective and safe at small doses for preventing or treating postoperative nausea, has been shown (at larger doses) to cause QT-interval lengthening and increase risk of Torsades de Pointes in a small cohort of susceptible patients. Therefore, a larger number of patients will be deprived of this useful medication because of our inability to identify the small number of patients who have the genetic markers associated with this rare but disastrous complication. Industry and academia are rapidly progressing toward simple assay-based genetic screens capable of identifying patients with these and other risks for adverse or inadequate drug responses, including heparin or warfarin resistance. Unfortunately, at the present time, commercially available screens are rare and have not been sufficiently evaluated to be considered the standard care. As such, detailed family history is the only practical means through which we can identify such genetic risks. At the same time, genetic variations may occur spontaneously or be present in a family but not be manifested with symptoms (phenotypically silent). Hence, continuous monitoring for adverse or highly variable drug responses, particularly those related to arrhythmias or BP instability, is the cornerstone of cardiovascular management in the perioperative period.
IV. Guidelines for prevention and treatment of cardiovascular disease
Drug treatment and drug prevention for several common cardiovascular diseases have been described in clinical practice guidelines published by national and international organizations. We provide references for the convenience of our readers. Because these recommendations evolve from year to year, we strongly recommend that readers check whether these guidelines may have been updated since publication of this volume.
A. CAD
1. Primary prevention (see Ref. 7)
2. Established disease (see Refs. 8,9)
3. Preoperative cardiac evaluation and prophylaxis during major surgery (see Refs. 10,11)
B. CHF (see Refs. 12,13)
C. Hypertension (see Refs. 14,15)
D. Atrial fibrillation prophylaxis (see Ref. 11)
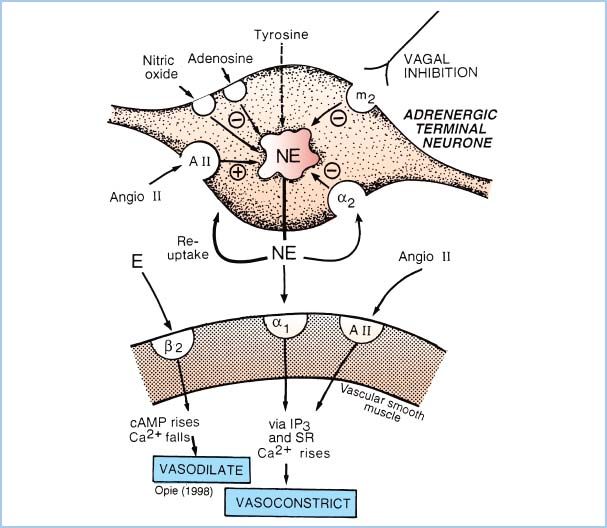
Figure 2.1 Schematic representation of the adrenergic receptors present on the sympathetic nerve terminal and vascular smooth-muscle cell. NE is released by electrical depolarization of the nerve terminal; however, the quantity of NE release is increased by neuronal (presynaptic) β2-receptor or muscarinic-cholinergic stimulation and is decreased by activation of presynaptic α2-receptors. On the postsynaptic membrane, stimulation of α1– or α2-adrenergic receptors causes vasoconstriction, whereas β2-receptor activation causes vasodilation. Prazosin is a selective α1-antagonist drug. Note that NE at clinical concentrations does not stimulate β2-receptors, but epinephrine (E) does. (From Opie LH. The Heart: Physiology from the Cell to the Circulation. Philadelphia, PA: Lippincott Williams & Wilkins; 1998:17–41, with permission.)
V. Vasopressors
A. a-Adrenergic receptor pharmacology (Fig. 2.1)
1. Postsynaptic α-adrenergic receptors mediate peripheral vasoconstriction (both arterial and venous), especially with neurally released norepinephrine (NE). Selective activation of cardiac α-adrenergic receptors increases inotropy while decreasing the HR. (Positive inotropy from α-adrenergic agonists can only be demonstrated in vitro or by selective drug administration in coronary arteries to avoid peripheral effects that normally overwhelm the cardiac actions.)
2. α-Adrenergic receptors on presynaptic nerve terminals decrease NE release through negative feedback. Activation of brain α-adrenergic receptors (e.g., with clonidine) lowers BP by decreasing sympathetic nervous system activity and causes sedation (e.g., with dexmedetomidine). Postsynaptic α2-adrenergic receptors mediate constriction of vascular smooth muscle.
3. Drug interactions
a. Reserpine interactions. Reserpine depletes intraneuronal NE and chronic use induces a “denervation hypersensitivity” state. Indirect-acting sympathomimetic drugs show diminished effect because of depleted NE stores, whereas direct-acting or mixed-action drugs may produce exaggerated responses because of receptor upregulation. This is of greater laboratory than clinical interest because of the rarity with which reserpine is now prescribed to patients. For the rare patient receiving reserpine, we recommend titrated dosages of direct-acting drugs and careful monitoring of BP.
b. Tricyclic (and tetracyclic) antidepressant or cocaine interactions. These drugs block the reuptake of catecholamines by prejunctional neurons and increase the catecholamine concentration at receptors. Interactions between these drugs and sympathomimetic agents can be very severe and of comparable or greater danger than the widely feared monoamine oxidase (MAO) inhibitor reactions. In general, if sympathomimetic drugs are required, small dosages of direct-acting agents represent the best choice.
4. Specific agents
a. Selective agonists
(1) Methoxamine (Vasoxyl)
(a) Methoxamine is a synthetic drug and is not a catecholamine. It is of mostly historic interest.
(b) Actions. The drug is a selective, direct, α1 agonist that produces vasoconstriction.
(c) Offset. A longer duration of action (1 to 1.5 h intramuscularly [IM]) than phenylephrine or NE; not metabolized by either MAO or catechol-O-methyl transferase (COMT).
(d) Indications for use—similar to phenylephrine
(e) Clinical use
(i) Methoxamine dose (adult): 1 to 5 mg intravenous (IV) bolus; 10 to 20 mg IM.
(ii) The long duration of action of methoxamine makes it more difficult to titrate the dosage to rapidly changing hemodynamic conditions than with phenylephrine, vasopressin, or NE.
(2) Phenylephrine (Neo-Synephrine) [16]
(a) Phenylephrine is a synthetic noncatecholamine.
(b) Actions. The drug is a selective α1-adrenergic agonist with minimal β-adrenergic effects. It causes vasoconstriction, primarily in arterioles.
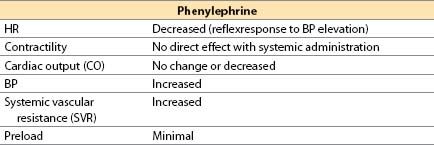
(c) Offset occurs by redistribution and rapid metabolism by MAO; there is no COMT metabolism.
(d) Advantages. A direct agonist with short duration (less than 5 min), it increases perfusion pressure for the brain, kidney, and heart in the presence of low SVR states. When used during hypotension, phenylephrine will increase coronary perfusion pressure without altering myocardial contractility. If hypertension is avoided, myocardial oxygen consumption (MVO2) does not increase substantially. If contractility is depressed because of ischemia, phenylephrine will sometimes produce an increased CO from an increase in coronary perfusion pressure. It is useful for correcting hypotension in patients with CAD, hypertrophic subaortic stenosis, tetralogy of Fallot, or valvular aortic stenosis.
(e) Disadvantages. It may decrease stroke volume (SV) and CO secondary to increased afterload; it may increase pulmonary vascular resistance (PVR); it may decrease renal, mesenteric, and extremity perfusion. Reflex bradycardia, usually not severe, may occur but will usually respond to atropine. Phenylephrine rarely may be associated with coronary artery spasm or spasm of an internal mammary, radial, or gastroepiploic artery bypass graft.
(f) Indications for use
(i) Hypotension due to peripheral vasodilation, low SVR states (e.g., septic shock, or to counteract effects of nitroglycerine)
(ii) For patients with supraventricular tachycardia (SVT), reflex vagal stimulation in response to increased BP may terminate the arrhythmia; phenylephrine treats both the hypotension and the arrhythmia.
2
(iii) It can oppose right-to-left shunting during acute cyanotic spells in tetralogy of Fallot.
(iv) Temporary therapy of hypovolemia until blood volume is restored, although a drug with positive inotropic action (e.g., ephedrine) usually is a better choice in patients without CAD (or hypertrophic obstructive cardiomyopathy), and in general, vasoconstrictors should not be viewed as effective treatments for hypovolemia.
(g) Administration. IV infusion (central line preferable) or IV bolus
(h) Clinical use
(i) Phenylephrine dose
(a) IV infusion: 0.5 to 10 μg/kg/min.
(b) IV bolus: 1 to 10 μg/kg, increased as needed (some patients with peripheral vascular collapse may require larger bolus injections to raise SVR).
(c) For tetralogy of Fallot spells in children: 5 to 50 μg/kg IV as a bolus dose.
(ii) Mixing:
(a) IV infusion: Often mix 10 or 15 mg in 250 mL IV fluid (40 or 60 μg/mL).
(b) IV bolus: Dilute to 40 to 100 μg/mL.
(iii) Nitroglycerin may be administered while maintaining or increasing arterial BP with phenylephrine. This combination may serve to increase myocardial oxygen supply while minimizing increases in MVO2.
(iv) Phenylephrine is the vasopressor of choice for short-term correction of excessive vasodilation in most patients with CAD or aortic stenosis.
b. Mixed agonists
(1) Dopamine (Intropin) [16]
See subsequent Section VI: Positive inotropic drugs.
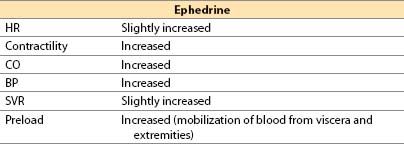
(2) Ephedrine
(a) Ephedrine is a plant-derived alkaloid with sympathomimetic effects.
(b) Actions
(i) Mild direct α-, β1-, and β2-adrenergic agonist
(ii) Indirect NE release from neurons
(c) Offset. 5 to 10 min IV; no metabolism by MAO or COMT; renal elimination
(d) Advantages
(i) Easily titrated pressor and inotrope that rarely produces unexpected excessive responses
(ii) Short duration of action with IV administration (3 to 10 min); lasts up to 1 h with IM administration
(iii) Limited tendency to produce tachycardia
(iv) Does not reduce blood flow to placenta; safe in pregnancy
(v) Good agent to correct sympathectomy-induced relative hypovolemia and decreased SVR after spinal or epidural anesthesia
(e) Disadvantages
(i) Efficacy is reduced when NE stores are depleted.
(ii) Risk of malignant hypertension with MAO inhibitors or cocaine
(iii) Tachyphylaxis with repeated doses (thus rarely administered by continuous infusion)
(f) Indications
(i) Hypotension due to low SVR or low CO, especially if HR is low, and particularly with spinal or epidural anesthesia
(ii) Temporary therapy of hypovolemia until circulating blood volume is restored, although, as previously noted, in general vasoconstrictors should not substitute for definitive treatment of hypovolemia
(g) Administration. IV, IM, subcutaneous (SC), by mouth (PO)
(h) Clinical use
(i) Ephedrine dose: 5 to 10 mg IV bolus, repeated or increased as needed; 25 to 50 mg IM.
(ii) Ephedrine is conveniently mixed in a syringe (5 to 10 mg/mL) and can be given as an IV bolus into a freely running IV line.
(iii) Ephedrine is a useful, quick-acting, titratable IV pressor that can be administered via a peripheral vein during anesthesia.
(3) Epinephrine (Adrenaline)
See subsequent Section IV: Positive inotropic drugs.
(4) NE (noradrenaline, Levophed) [16]
(a) NE is the primary physiologic postganglionic sympathetic neurotransmitter; NE also is released by adrenal medulla and central nervous system (CNS) neurons.
(b) Actions
(i) Direct α1– and α2-adrenergic actions and β1-adrenergic agonist action
(ii) Limited β2-adrenergic effect in vivo, despite NE being a more powerful β2-adrenergic agonist than dobutamine in vitro
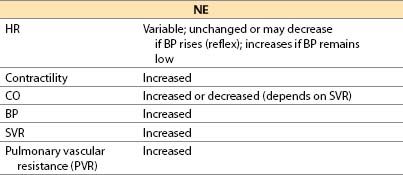
(c) Offset is by redistribution, neural uptake, and metabolism by MAO and COMT.
(d) Advantages
(i) Direct adrenergic agonist. Equipotent to epinephrine at β1-adrenergic receptors
(ii) Redistributes blood flow to brain and heart because all other vascular beds are constricted.
(iii) Elicits intense α1– and α2-adrenergic agonism; may be effective as vasoconstrictor when phenylephrine (α1 only) lacks efficacy
(e) Disadvantages
(i) Reduced organ perfusion: Risk of ischemia of kidney, skin, liver, bowel, and extremities
(ii) Myocardial ischemia possible; increased afterload, HR. Contractility may increase, be unchanged, or even decrease. Coronary spasm may be precipitated.
(iii) Pulmonary vasoconstriction
(iv) Arrhythmias
(v) Risk of skin necrosis with SC extravasation
(f) Indications for use
(i) Peripheral vascular collapse when it is necessary to increase SVR (e.g., septic shock or “vasoplegia” after cardiopulmonary bypass [CPB])
(ii) Conditions in which increased SVR is desired together with cardiac stimulation
(iii) Need for increased SVR in which phenylephrine has proved ineffective
(g) Administration. IV only, by central line only
(h) Clinical use
(i) Usual NE starting infusion doses: 15 to 30 ng/kg/min IV (adult); usual range, 30 to 300 ng/kg/min.
(ii) Minimize duration of use; monitor patient for oliguria and metabolic acidosis.
(iii) NE can be used with vasodilator (e.g., nitroprusside or phentolamine) to counteract α stimulation while leaving β1-adrenergic stimulation intact; however, if intense vasoconstriction is not required, we recommend that a different drug be used.
(iv) For treating severe right ventricular (RV) failure associated with cardiac surgery, the simultaneous infusion of NE into the left atrium (through a left atrial catheter placed intraoperatively) plus inhaled NO and/or nitroglycerine by IV infusion is useful. The left atrial NE reaches the systemic vascular bed first and it is largely metabolized peripherally before it reaches the lung where it might increase PVR (had the NE been infused through a venous line) (Table 2.2).
(5) Interactions with MAO inhibitors
(a) MAO is an enzyme that deaminates NE, dopamine, and serotonin. Thus, the MAO inhibitors treat severe depression by increasing catecholamine concentrations in the brain by inhibiting catecholamine breakdown. Administration of indirectly acting adrenergic agonists or meperidine to patients taking MAO inhibitors can produce a life-threatening hypertensive crisis. Ideally, 2 to 3 wks should elapse between discontinuing the hydrazine MAO inhibitor phenelzine and elective surgery. Nonhydrazine MAO inhibitors (isocarboxazid, tranylcypromine) require 3 to 10 days for offset of effect. Selegiline at doses 10 mg or less per day should present fewer adverse drug interactions than other MAO inhibitors.
(b) The greatest risk of inducing a hyperadrenergic state occurs with indirect- acting sympathomimetic drugs (such as ephedrine), because such agents release the intraneuronal stores of NE that were increased by the MAO inhibitor. Because dopamine releases NE, it should be initiated with caution in the MAO-inhibited patient.
Table 2.2 Acute treatment of pulmonary hypertension and RV failure
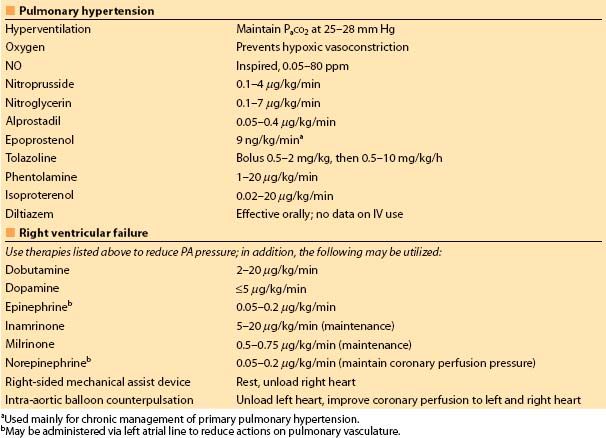
(c) In MAO-inhibited patients, preferred drugs are those with purely direct activity: epinephrine, NE, isoproterenol, phenylephrine, vasopressin, and dobutamine. All pressor drugs should be used cautiously, in small dosages with BP monitoring and observation of the electrocardiogram (ECG) for arrhythmias.
B. Vasopressin pharmacology and agonists [17]
1. Actions
a. Vasopressin is an endogenous antidiuretic hormone that in high concentrations produces a direct peripheral vasoconstriction through activation of smooth-muscle V1 receptors. Vasopressin has no actions on β-adrenergic receptors, so it may produce less tachycardia than epinephrine when used for resuscitation after cardiac arrest. Vasopressin has been administered intra-arterially in a selective fashion to control gastrointestinal bleeding.
b. Vasopressin produces relatively more vasoconstriction of skin, skeletal muscle, intestine, and adipose tissue than of coronary or renal beds. Vasopressin causes cerebrovascular dilation.
2. Advantages
a. Vasopressin is a very potent agent which acts independently of adrenergic receptors.
b. Some studies suggest that vasopressin may be effective at maintaining adequate SVR in severe acidosis, sepsis, or after CPB, even when agents such as phenylephrine or NE have proven ineffective.
c. Vasopressin may restore coronary perfusion pressure after cardiac arrest without also producing tachycardia and arrhythmias, as is common when epinephrine is used for this purpose.
3. Disadvantages
a. Vasopressin produces a variety of unpleasant signs and symptoms in awake patients, including pallor of skin, nausea, abdominal cramping, bronchoconstriction, and uterine contractions.
b. Decreases in splanchnic blood flow may be of concern in patients receiving vasopressin for more than a few hours, particularly when vasopressin is coadministered with agents such as α-adrenergic agonists and positive inotropic drugs. Increases in serum concentrations of bilirubin and of “liver” enzymes are common.
c. Vasopressin may be associated with a decrease in platelet concentration.
d. Lactic acidosis is common in patients receiving vasopressin infusion (but these patients are generally critically ill and already receiving other vasoactive agents).
4. Clinical use
a. Vasopressin has been used as an alternative to epinephrine in treating countershock-refractory ventricular fibrillation (VF) in adults. The 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science suggest that vasopressin may be substituted either for the first or second dose of epinephrine during resuscitation, but that there is no convincing evidence that inclusion of vasopressin in resuscitation will improve outcomes relative to outcomes obtained with epinephrine alone. The typical vasopressin resuscitation dose is 40 units as an IV bolus.
b. Vasopressin has been used in a variety of conditions associated with vasodilatory shock, including sepsis, the “vasoplegic” syndrome after CPB, and for hypotension occurring in patients receiving ACE inhibitors (or angtiotensin receptor blockers) and general anesthesia. Typical adult doses range from 4 to 6 units/h. We have found this drug to be effective and useful, but sometimes associated with troublesome metabolic acidosis. We speculate that the latter may be the result of inadequate visceral perfusion.
VI. Positive inotropic drugs
A. Treatment of low CO [16]
1. Goals. Increase organ perfusion and oxygen delivery to tissues
a. Increase CO by increasing SV, HR (when appropriate) or both
b. Maximize myocardial oxygen supply (increase diastolic arterial pressure, diastolic perfusion time, and blood O2 content; decrease left ventricular end-diastolic pressure [LVEDP])
c. Provide an adequate mean arterial pressure (MAP) for perfusion of other organs
d. Minimize increases in myocardial oxygen demand by avoiding tachycardia and left ventricular (LV) dilation
e. Metabolic disturbances, arrhythmias, or cardiac ischemia, if present, should be treated concurrently.
f. Drug treatment of critically ill patients with intrinsic myocardial failure may include the following cardiac stimulants:
(1) β1-Adrenergic stimulation
(2) Phosphodiesterase (PDE) inhibition
(3) Dopaminergic stimulants
(4) Calcium sensitizers (increase calcium sensitivity of contractile proteins)
(5) Digoxin
2. Monitoring. Positive inotropic drug dosing is most effectively regulated using data from the arterial line, a monitor of cardiac output, and/or echocardiography. In addition, monitoring of mixed venous oxygen saturation can be extremely valuable. The inotropic drug dosage can be titrated to CO and BP endpoints, together with assessment of organ perfusion, e.g., urine output and concentration.
B. cAMP-dependent agents
1. b-Adrenergic and dopaminergic receptor agonists
a. Similarities among sympathomimetic drugs
(1) β1-agonist effects are primarily stimulatory.
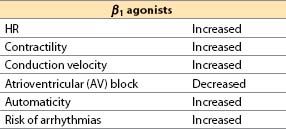
(2) β2 agonists cause vasodilation and bronchodilation, and they also increase HR and contractility (albeit with less potency than β1 agonists).
(3) Postsynaptic dopaminergic receptors mediate renal and mesenteric vasodilation, increase renal salt excretion, and reduce gastrointestinal motility. Presynaptic dopaminergic receptors inhibit NE release.
(4) Diastolic ventricular dysfunction. Cardiac β-receptor activation enhances diastolic ventricular relaxation by facilitating the active, energy-consuming process that pumps free intracellular Ca2+ into storage sites. Abnormal relaxation occurring in ischemia and other myocardial disorders leads to increased diastolic stiffness. By augmenting diastolic relaxation, β-adrenergic receptor agonists reduce LVEDP and heart size (LV end-diastolic volume [LVEDV]), improve diastolic filling, reduce left atrial pressure (LAP), and improve the myocardial oxygen supply–demand ratio.
(5) Systolic ventricular dysfunction. More complete ventricular ejection during systole will reduce the LV end-systolic volume. This reduces heart size, LV systolic wall tension (by Laplace’s law), and myocardial oxygen consumption (MVO2).
(6) Myocardial ischemia. The net effects of β-receptor stimulation on myocardial O2 supply and demand are multifactorial and may be difficult to predict. MVO2 tends to increase as HR and contractility rise, but MVO2 is reduced by lowering LVEDV. β Agonists improve O2 supply when LVEDP is decreased, but can worsen the supply–demand ratio particularly if HR rises or diastolic BP is lowered.
(7) Hypovolemia is deleterious to the patient with heart failure just as it is for the patient with normal ventricular function; however, volume overload may lead to myocardial ischemia by restricting subendocardial perfusion.
(8) There is a risk of tissue damage or sloughing when vasoactive drugs extravasate outside of a vein. In general, catecholamine vasoconstrictors should not be infused for long periods of time through a peripheral IV line because of the risk of extravasation or infiltration. These drugs may be given through peripheral IV lines provided that
(a) No central venous catheter is available.
(b) The drug is injected only into a free-flowing IV line.
(c) The IV site is observed during and after the injection for signs of infiltration or extravasation.
b. Dobutamine (Dobutrex) [16]
(1) Dobutamine is a synthetic catecholamine formulated as a racemic mixture.
(2) Actions
(a) Direct β1 agonist, with limited β2 and α1 effects. Dobutamine has no α2 or dopaminergic activity.
(b) Dobutamine increases cardiac inotropy principally via its β1 (and perhaps also by α1) agonism, but HR is increased only by the β1 effect.
(c) On blood vessels, dobutamine is predominantly a vasodilator drug. Mechanisms for vasodilation include the following:
(i) β2-mediated vasodilation that is only partially counteracted by (−) dobutamine’s α1 constrictor effects
(ii) The (+) dobutamine enantiomer and its metabolite, (+)-3-O-methyldobutamine, are α1 antagonists. Thus, as dobutamine is metabolized, any α1 agonist actions of the drug should diminish over time.
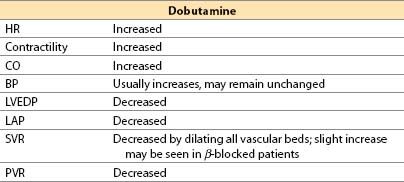
(3) Offset. Offset of action is achieved by redistribution, metabolism by COMT, and conjugation by glucuronide in liver; an active metabolite is generated. Plasma half-life is 2 min.
(4) Advantages
(a) At low doses there is generally less tachycardia than with “equivalently inotropic” doses of isoproterenol or dopamine, whereas some studies show that “equivalently inotropic” doses of epinephrine produce LESS tachycardia than dobutamine.
(b) Afterload reduction (SVR and PVR) may improve LV and RV systolic function, which can benefit the heart with right and/or LV failure.
(c) Renal blood flow may increase (due to a β2 effect), but not as much as with comparable (but low) doses of dopamine or dopexamine.
(5) Disadvantages
(a) Tachycardia and arrhythmias are dose-related and can be severe.
(b) Hypotension may occur if the reduction in SVR is not fully offset by an increase in CO; dobutamine is an inotrope but is not a pressor.
(c) Coronary steal is possible.
(d) The drug is a nonselective vasodilator: Blood flow may be shunted from kidney and splanchnic bed to skeletal muscle.
(e) Tachyphylaxis has been reported when infused for more than 72 h.
(f) Mild hypokalemia may occur.
(g) As a partial agonist, dobutamine can inhibit actions of full agonists (e.g., epinephrine) under certain circumstances.
(6) Indications. Low CO states, especially with increased SVR or PVR
(7) Administration. IV only (central line is preferable, but dobutamine has little vasoconstrictor activity, minimizing risk of extravasation).
(8) Clinical use
(a) Dobutamine dose: IV infusion, 2 to 20 μg/kg/min. Some patients may respond to initial doses as low as 0.5 μg/kg/min and, at such low doses, HR usually does not increase.
(b) Dobutamine increases MVO2 to a lesser degree than CO. Dobutamine increases coronary blood flow to a greater degree than dopamine when either agent is given as a single drug. However, addition of nitroglycerin to dopamine may be equally effective.
(c) Dobutamine acts similar to a fixed-ratio combination of an inotropic drug and a vasodilator drug. These two components cannot be titrated separately.
(d) In patients undergoing coronary surgery, dobutamine produces more tachycardia than epinephrine when administered to produce the same increase in SV [11].
(e) When dobutamine is given to β-blocked patients, SVR may increase.
(f) Routine administration of dobutamine (or any other positive inotrope) is not recommended. [18]
c. Dopamine (Intropin) [16]
(1) Dopamine is a catecholamine precursor to NE and epinephrine found in nerve terminals and the adrenal medulla.
(2) Actions
(a) Direct action: α1-, β1-, β2-adrenergic, and dopaminergic (DA1) agonist
(b) Indirect action: Induces release of stored neuronal NE
(c) The dose versus response relationship is often described as if “carved in stone”; however, the relationship between dose and concentration and between dose and response is highly variable from patient to patient.
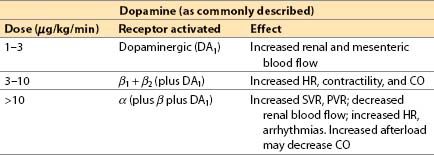
(3) Offset is achieved by redistribution, uptake by nerve terminals plus metabolism by MAO and COMT.
(4) Advantages
(a) Increased renal perfusion and urine output at low to moderate dosages (partially due to a specific DA1 agonist effect)
(b) Blood flow shifts from skeletal muscle to kidney and splanchnic beds.
(c) BP response is easy to titrate because of its mixed inotropic and vasoconstrictor properties.
(5) Disadvantages
(a) There is a significant indirect-acting component; response can diminish when neuronal NE is depleted (e.g., in patients with CHF).
(b) Sinus, atrial, or ventricular tachycardia (VT) and arrhythmias may occur.
(c) Maximal inotropic effect less than that of epinephrine.
(d) Skin necrosis may result from extravasation.
(e) Renal vasodilating effects are over-ridden by α-mediated vasoconstriction at dosages greater than 10 μg/kg/min with risk of renal, splanchnic, and skin necrosis. Urine output should be monitored.
(f) Pulmonary vasoconstriction is possible.
(g) MVO2 increases, and myocardial ischemia may occur if coronary flow does not increase commensurately.
(h) In some patients with severe HF, the increased BP at increased doses may be detrimental. Such patients benefit from adding a vasodilator.
(6) Indications
(a) Hypotension due to low CO or low SVR (although other agents are superior for the latter indication)
(b) Temporary therapy of hypovolemia until circulating blood volume is restored (but vasoconstrictors should not substitute or delay primary treatment of hypovolemia)
(c) “Recruiting renal blood flow” for renal failure or insufficiency (widely used for this purpose, but limited evidence basis)
(7) Administration: IV only (preferably by central venous line)
(8) Clinical use
(a) Dopamine dose: 1 to 20 μg/kg/min IV.
(b) Often mix 200 mg in 250 mL IV solution (800 μg/mL).
(c) Good first choice for temporary treatment of hypotension until intravascular volume can be expanded or until a specific diagnosis can be made.
(d) Correct hypovolemia if possible before use (as with all pressors)
(e) After cardiac surgery if inotropic response is not adequate at dopamine doses of 5 to 10 μg/kg/min, we recommend a switch to a more powerful agonist such as epinephrine, or a switch to or addition of milrinone.
(f) Consider adding a vasodilator (e.g., nitroprusside) when BP is adequate and afterload reduction would be beneficial (or better still, reduce the dose of dopamine).
d. Dopexamine [16]
(1) Actions
(a) Dopexamine is a synthetic analog of dobutamine with vasodilator action. Its cardiac inotropic and chronotropic activity is caused by direct β2-agonist effects and by NE actions (due to baroreceptor reflex activation and neuronal uptake-1 inhibition) that indirectly activate β1-receptors. In CHF, there is selective β1 downregulation, with relative preservation of β2-receptor number and coupling. The latter assume greater than normal physiologic importance, making dopexamine of theoretically greater utility than agents with primary β1-receptor activity. Although dopexamine has been used in Europe for roughly a decade, the drug will likely never be available in the United States.
(b) Receptor activity
α1 and α2: Minimal
β1: Little direct effect, some indirect; β2: Direct agonist
DA1: Potent agonist (activation increases renal blood flow)
(c) Inhibits neuronal catecholamine uptake-1, increasing NE actions
(d) Hemodynamic actions
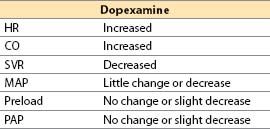
(2) Offset. Half-life: 6 to 11 min. Clearance is by uptake into tissues (catecholamine uptake mechanisms) and by hepatic metabolism.
(3) Advantages
(a) Lack of vasoconstrictor activity avoids α-mediated complications.
(b) Decreased renovascular resistance might theoretically help preserve renal function after ischemic insults.
(4) Disadvantages
(a) Less effective positive inotrope than other agents (e.g., epinephrine, milrinone)
(b) Dose-dependent tachycardia may limit therapy.
(c) Tachyphylaxis
(d) Not approved by the U.S. Food and Drug administration for release in the United States.
(5) Indications. Treatment of low CO states
(6) Administration. IV
(7) Clinical use
(a) Dopexamine dose: 0.5 to 4 μg/kg/min IV (maximum 6 μg/kg/min).
(b) Hemodynamic and renal effects similar to the combination of variable doses of dobutamine with dopamine 1 μg/kg/min (renal dose) or fenoldopam 0.05 μg/kg/min.
e. Epinephrine (Adrenaline) [16]
(1) Epinephrine is a catecholamine produced by the adrenal medulla.
(2) Actions
(a) Direct agonist at α1-, α2-, β1-, and β2-receptors
(b) Dose response (adult, approximate)
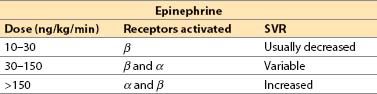
(c) Increased contractility with all dosages, but SVR may decrease, remain unchanged, or increase dramatically depending on the dosage. CO usually increased but, at extreme resuscitation dosages, α-receptor–mediated vasoconstriction may cause a lowered SV due to high afterload.
(3) Offset occurs by uptake by neurons and tissue and by metabolism by MAO and COMT (rapid).
(4) Advantages
(a) This drug is direct-acting; its effects are not dependent on release of endogenous NE.
(b) Potent α– and β-adrenergic stimulation results in greater maximal effects and produce equivalent increases in SV with tachycardia after heart surgery than dopamine or dobutamine.
(c) It is a powerful inotrope with variable (and dose-dependent) α-adrenergic effect. Lusitropic effect (β1) enhances the rate of ventricular relaxation.
(d) BP increases may blunt tachycardia due to reflex vagal stimulation.
(e) It is an effective bronchodilator and mast cell stabilizer, useful for primary therapy of severe bronchospasm, anaphylactoid, or anaphylactic reactions.
(f) With a dilated LV and myocardial ischemia, epinephrine may increase diastolic BP and decrease heart size, reducing myocardial ischemia. However, as with any inotropic drug, epinephrine may induce or worsen myocardial ischemia.
3
(5) Disadvantages
(a) Tachycardia and arrhythmias at higher doses.
(b) Organ ischemia, especially kidney, secondary to vasoconstriction, may result. Renal function must be closely monitored.
(c) Pulmonary vasoconstriction may occur, which can produce pulmonary hypertension and possibly RV failure; addition of a vasodilator may counteract this.
(d) Epinephrine may produce myocardial ischemia. Positive inotropy and tachycardia increase myocardial oxygen demand and reduce oxygen supply.
(e) Extravasation from a peripheral IV cannula can cause necrosis; thus, administration via a central venous line is preferable.
(f) As with most adrenergic agonists, increases of plasma glucose and lactate occur. This may be accentuated in diabetics.
(g) Initial increases in plasma K+ occur due to hepatic release, followed by decreased K+ due to skeletal muscle uptake.
(6) Indications
(a) Cardiac arrest (especially asystole or VF); electromechanical dissociation. Epinephrine’s efficacy is believed to result from increased coronary perfusion pressure during cardiopulmonary resuscitation (CPR). Recently, the utility of high-dose (0.2 mg/kg) epinephrine was debated, the consensus is that there is no outcome benefit to “high-dose” epinephrine.
(b) Anaphylaxis and other systemic allergic reactions; epinephrine is the agent of choice.
(c) Cardiogenic shock, especially if a vasodilator is added
(d) Bronchospasm
(e) Reduced CO after CPB
(f) Hypotension with spinal or epidural anesthesia can be treated with low-dose (1 to 4 μg/min) epinephrine infusions as conveniently and effectively as with ephedrine boluses [12].
(7) Administration. IV (preferably by central line); via endotracheal tube (rapidly absorbed by tracheal mucosa); SC
(8) Clinical use
(a) Epinephrine dose
(i) SC: 10 μg/kg (maximum of 400 μg or 0.4 mL, 1:1,000) for treatment of mild-to-moderate allergic reactions or bronchospasm.
(ii) IV: Low-to-moderate dose (for shock, hypotension): 0.03 to 0.2 μg/kg bolus (IV), then infusion at 0.01 to 0.30 μg/kg/min.
High dose (for cardiac arrest, resuscitation): 0.5 to 1.0 mg IV bolus; pediatric, 5 to 15 μg/kg (may be given intratracheally in 1 to 10 mL volume). Larger doses are used when response to initial dose is inadequate.
Resuscitation doses of epinephrine may produce extreme hypertension, stroke, or myocardial infarction. A starting dose of epinephrine exceeding a 150 ng/kg (10 μg in an adult) IV bolus should be given only to a patient in extremis! Moderate doses (.03–.06 μg/kg/min) of epinephrine are commonly used to stimulate cardiac function an facilitate separation from cardiopulmonary bypass.
(b) Watch for signs of excessive vasoconstriction. Monitor SVR, renal function, extremity perfusion.
(c) Addition of a vasodilator (e.g., nicardipine, nitroprusside, or phentolamine) to epinephrine can counteract the α-mediated vasoconstriction, leaving positive cardiac inotropic effects undiminished. Alternatively, addition of milrinone or inamrinone may permit lower doses of epinephrine to be used. We find combinations of epinephrine and milrinone particularly useful in cardiac surgical patients.
f. NE (noradrenaline, Levophed)
See the preceding Section V: Vasopressors.
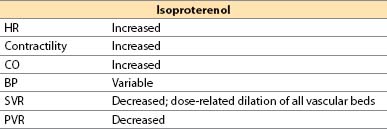
g. Isoproterenol (Isuprel)
(1) Isoproterenol is a synthetic catecholamine.
(2) Actions
(a) Direct β1– plus β2-adrenergic agonist
(b) No α-adrenergic effects
Related posts:
 Devices for Cardiac Support and Replacement
Devices for Cardiac Support and Replacement
 Alternative Approaches to Cardiac Surgery with and without Cardiopulmonary Bypass
Alternative Approaches to Cardiac Surgery with and without Cardiopulmonary Bypass
 Anesthetic Considerations for Patients with Pericardial Disease
Anesthetic Considerations for Patients with Pericardial Disease
 Devices for Cardiac Support and Replacement
Devices for Cardiac Support and Replacement
 Anesthetic Management for Patients with Congenital Heart Disease: The Adult Population
Anesthetic Management for Patients with Congenital Heart Disease: The Adult Population
 Cardiovascular Drugs
Cardiovascular Drugs
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


