INTRODUCTION
β-Adrenergic receptor antagonists (β-blockers) are medications used in the treatment of various cardiovascular, neurologic, endocrine, ophthalmologic, and psychiatric disorders. Among all the exposures to cardiovascular agents, β-blocker exposures were the leading cause of poison center calls and ranked among the top three in this class as a cause of severe toxicity and mortality.1
PHARMACOLOGY
The β-adrenergic receptors are membrane glycoproteins present as three subtypes in various tissues (Table 194-1). These receptors play a critical role in cardiovascular physiology by modulating cardiac activity and vascular tone.
| β-Receptor Type | Location | Agonism | Antagonism |
|---|---|---|---|
| β1 | Myocardium Kidney Eye | Increases inotropy Increases chronotropy Stimulates renin release Stimulates aqueous humor production | Decreases inotropy Decreases chronotropy Inhibits renin release Inhibits aqueous humor production |
| β2 | Bronchial smooth muscle Visceral smooth muscle Skeletal muscle Liver Vascular | Causes bronchodilation Relaxes uterus Causes ileus Increases force of contraction Stimulates glycogenolysis Stimulates glycogenolysis and gluconeogenesis Vasodilation | Causes bronchospasm — — Inhibits glycogenolysis and gluconeogenesis Minimal vasoconstriction |
| β3 | Adipose tissue Skeletal muscle | Stimulates lipolysis Stimulates thermogenesis | Inhibits lipolysis Inhibits thermogenesis |
During times of stress (i.e., catecholamine release), β-adrenergic receptor stimulation increases myocardial and vascular smooth muscle cell activity through a sequence of intracellular events (Figure 194-1).2,3
FIGURE 194-1.
Cardiac myocyte β1-receptor and calcium signaling. Following myocyte depolarization, extracellular calcium (Ca2+) enters the cell via the L-type or voltage-gated calcium channel (L-VDCC) and binds to the ryanodine receptor (RyR) in the sarcoplasmic reticulum, causing an efflux of sequestered Ca2+ out of the sarcoplasmic reticulum into the cytosol. Free Ca2+ binds to troponin that allows the myosin and actin interaction, resulting in contraction of the cardiac myocyte. Binding of a β-agonist to the β1-adrenergic receptor (B1) on the cell surface activates the Gs protein. The Gs protein then activates adenylate cyclase (AC), which converts adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP). The increased cAMP activates protein kinase A (PKA). Activated PKA serves as further stimulus for the L-VDCC opening. Glucagon independently activates adenylate cyclase. cAMP is metabolized by phosphodiesterase (PDE) into inactive adenosine 5′-monophosphate (5’AMP).
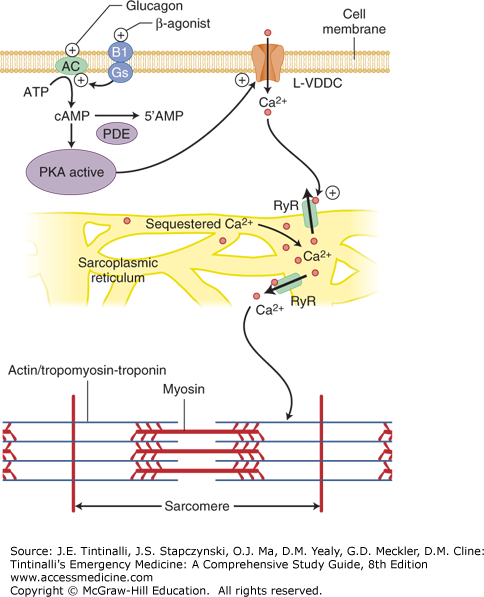
The β-receptor is coupled to a stimulatory Gs protein. This Gs protein stimulates adenylate cyclase, which in turn catalyzes the formation of cyclic adenosine monophosphate, the so-called intracellular second messenger. Increased cyclic adenosine monophosphate ultimately phosphorylates the L-type calcium channel, which leads to channel opening and calcium entry into the cell. Extracellular calcium is then coupled to the ryanodine receptor to carry the calcium current to the sarcoplasmic reticulum, which then releases its stored calcium. This process is termed calcium-induced calcium release. Stored calcium becomes available to participate in mechanical contraction via the actin and myosin complex. Like the cardiac myocyte, the vascular smooth muscle uses L-type calcium channels to regulate intracellular calcium and subsequently coordinate vascular tone. To prevent overdrive of the cell, phosphodiesterase breaks down cyclic adenosine monophosphate to adenosine 5′-monophosphate, thus removing the stimulus for calcium channel opening, and the contractile process ceases.
The β-blockers modulate the activity of myocyte and vascular smooth muscle contraction by decreasing calcium entry into the cell.2,3 Therapeutically, β-blockade lessens the work performed by the diseased or injured myocardium and lowers elevated blood pressure. On the other hand, excessive β-blockade may lead to profound pump failure, with bradycardia, decreased contractility, and hypotension.2
The pharmacologic properties of various β-blockers influence their spectrum of action, adverse drug reactions, and toxicity (Table 194-2).4,5 These properties include receptor selectivity, sodium channel blockade (also known as membrane-stabilizing activity), lipid solubility, protein binding, and partial agonist activity (also known as intrinsic sympathomimetic activity). For example, highly lipid-soluble agents, such as propranolol, readily cross the blood–brain barrier and achieve high concentrations in brain tissue.2,3 This may contribute to the more severe CNS manifestations of mental status depression, seizures, and coma seen after an overdose of such agents.2,3 Several β-blockers inhibit myocardial sodium channels, similar to quinidine and cyclic antidepressants, rendering these drugs potentially more cardiodepressant following overdose.3 However, in massive overdoses, all β-blockers can be severely cardiodepressive.6
| Agent | β1 Selectivity | Lipophilicity | Partial Agonism | Protein Binding (%) | Sodium Channel Blockade | Half-Life (h) |
|---|---|---|---|---|---|---|
| Acebutolol | + | Moderate | + | 25 | + | 3–4 |
| Atenolol | + | Weak | 0 | 6–16 | 0 | 6–9 |
| Betaxolol | + | High | 0 | 55 | ± | 14–22 |
| Bisoprolol | ++ | Moderate | 0 | 30–40 | 0 | 9–12 |
| Carvedilol | 0 | Moderate | 0 | >95 | ± | 7–10 |
| Esmolol | + | Weak | 0 | 55 | ± | 9 min |
| Labetalol | + | Weak | 0 | 50 | ± | 3–4 |
| Metoprolol | ++ | Moderate | 0 | 12 | ± | 3–4 |
| Nadolol | 0 | Weak | 0 | 30 | 0 | 12–24 |
| Nebivolol | +++ | Moderate | 0 | 98 | 0 | 8–27 |
| Oxprenolol | 0 | Moderate | ++ | 80 | + | 1–2 |
| Pindolol | 0 | High | ++ | 40–60 | ± | 3–4 |
| Penbutolol | 0 | High | + | 80–98 | 0 | 5–20 |
| Propranolol | 0 | High | 0 | >90 | ++ | 3–4 |
| Sotalol | 0 | Weak | 0 | Minimal | 0 | 12 |
| Timolol | 0 | High | ± | 10–60 | 0 | 4–5 |
Although β1 cardioselective medications have less risk of unwanted β2 effects, such as bronchospasm, selectivity is often lost following large overdoses.3 Several β-blockers like pindolol have partial agonist activity, causing weak stimulation of the β-receptor, with a lessor tendency for bradycardia during therapeutic use.4 Some β-blockers, such as labetalol and carvedilol, are also antagonists at α1-adrenergic receptors, which can result in exaggerated hypotension during therapeutic use. Sotalol is unique among β-blockers in its ability to block potassium channels important for repolarization, as do other class III antiarrhythmic drugs.2,3,4
In addition to having cardiopulmonary effects, β-blockers also alter metabolism in the liver, skeletal muscle, and adipose tissue. Under normal conditions, the heart uses free fatty acids as its primary energy source, but during times of stress, it switches to using carbohydrates to maintain metabolism. Inhibition of glycogenolysis and gluconeogenesis reduces the availability of carbohydrates for use by metabolically active cells. Although hypoglycemia can occur as a consequence of β-blocker toxicity, it is actually uncommon.2 In the presence of adequate glucose stores, euglycemia and hyperglycemia are more common than hypoglycemia.
Clinically relevant pharmacokinetic characteristics include drug formulation (regular or extended release), rate of drug absorption, protein binding, lipid solubility, elimination mostly by hepatic metabolism, and volume of distribution. These properties determine onset of symptoms, duration of symptoms, target organ toxicity, and potential treatment modalities.
CLINICAL FEATURES
Toxicity due to β-blockers can produce a spectrum of clinical symptoms (Table 194-3).2,3,7 The timing of symptom appearance depends upon the formulation. Absorption of regular-release β-blockers occurs rapidly, often with peak effects within 1 to 4 hours. However, delays of up to 6 hours following acute ingestion have occurred.8 Experience is limited regarding onset of symptoms with poisoning following an ingestion of sustained-release β-blocker formulations, but based on other sustained-release cardiac drugs, it is assumed that symptoms may be delayed >6 hours after ingestion.2,3 Co-ingestants that alter gut function, such as opioids and anticholinergics, may affect absorption of β-blockers and subsequent onset of symptoms.2
Cardiovascular
CNS
Pulmonary
Electrolytes
|
The primary organ system affected by β-blocker toxicity is the cardiovascular system, and the hallmark of severe toxicity is bradycardia and shock.2,3,7,9 Bradycardia due to sinus node suppression or conduction abnormalities occurs in virtually all significant β-blocker intoxications, although ingestion of β-blockers with partial agonist activity may initially present with hypertension and tachycardia.9 The β-blockers with sodium channel antagonism can worsen conduction abnormalities, causing a wide-complex bradycardia (especially when the QRS interval is >100 milliseconds).9
The cardiotoxic profile of sotalol is different from that of other β-blockers due to its ability to block potassium channels and prolong the QT interval.3 Thus, sotalol is more often associated with ventricular dysrhythmias, including premature ventricular contractions, bigeminy, ventricular tachycardia, ventricular fibrillation, and torsades de pointes.3
β-Blockers also affect the CNS and pulmonary system. Neurologic manifestations include depressed mental status, coma, and seizures.2 These symptoms most likely occur as a result of a combination of hypoxia due to poor perfusion, sodium channel antagonism, and direct neuronal toxicity.2 More lipophilic β-blockers, such as propranolol, cause greater neurologic toxicity than the less lipophilic agents.9 Seizures are generally brief, and status epilepticus is rare.2 Nonselective β-blockers may antagonize the β2-receptor in bronchial smooth muscle causing bronchospasm. Similarly, in large ingestions of cardioselective β-blockers, the β1 selectivity may be lost.
DIAGNOSIS
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


