(1)
Northern Anesthesia & Pain Medicine, LLC, Eagle River, Alaska, USA
(2)
WWAMI Program, University of Washington School of Medicine, Anchorage, Alaska, USA
Keywords
OpioidVectorTransmissionBest practicesPainEvidence basedCartesianNociceptionTransductionAδDorsal hornSensitizationWindupModulationPerceptionGate theoryInterneuronNeuromatrixBiopsychosocialPreventionMultimodalSpiritualA gynecologist colleague requests that a thoracic epidural catheter be placed for perioperative pain management for a planned laparoscopic supracervical hysterectomy for a 32-year-old female suffering from intractable chronic pelvic pain thought to be caused by endometriosis which was diagnosed via prior laparoscopy. Thinking this an unusual request (epidurals are typically reserved for more painful operations with large incisions and especially supraumbilical incisions which may compromise diaphragmatic excursion by respiratory splinting), you probe for a little more detail. Your colleague simply says, “just go see her in Bay 4.”
When you arrive to assess the patient, there are concerned family members surrounding the bed of a somnolent and dysphoric young woman with pinpoint pupils. Upon further questioning, you ascertain that she has seen multiple physicians for complaints of chronic pelvic pain and was initially started on opioid therapy by her primary care physician who subsequently referred her to a local pain management clinic who has escalated her regimen to extended-release morphine sulfate 120 mg twice a day and hydrocodone-acetaminophen 10 mg/325 mg six per day. She is also using alprazolam as an anxiolytic and carisoprodol as a “muscle relaxant .”
After further evaluation and discussion, you agree to place a thoracic epidural catheter which is used to provide preemptive analgesia prior to incision and continued via infusion for the next 3 days while she is in the hospital. During this time, you also provide a morphine PCA with no basal rate and a total hourly lockout rate at 16 mg/h for the first day, transitioning to Percocet p.o. the second day when she is taking solids. Scheduled lorazepam and clonidine are also provided. She denies any abdominopelvic pain and consistently evidences bilateral sensory block from roughly T6 to L2 levels with an epidural infusion of ropivacaine . She suffers mild opioid withdrawal symptoms primarily consisting of tachycardia and some agitation during the first 2 days, but by the third day, she displays a dramatically improved mental status and bright affect and expresses gratitude for your care.
Two weeks later when you follow up with her by telephone, she is excited to report that she destroyed her remaining opioids at home and vows “never to take that stuff again!” She is also excited to report that she has a job interview.
Introduction
Transmission of an infectious agent frequently requires a vector, which entity serves as a reservoir for the agent, either symptomatically or asymptomatically. Common examples include anopheles mosquitoes carrying Plasmodium falciparum or deer ticks harboring Borrelia burgdorferi . Unique among epidemics to date, this agent (opioids) is “transmitted” primarily by those to whom the well-being of the individual and also the community is entrusted. As such, a thorough understanding of opioids is essential for every prescriber, and so we began with basic opioid biology and pharmacology in Chaps. 2, 3, and 4. Knowing about the drugs is not enough however; the prescriber must know why, when, and how to use them and, perhaps of greater importance, when not to. These specific questions are addressed in the two chapters that follow.
Altering “transmission ” of opioids via professional vectors is a laborious but not insurmountable task. Recent intensive provider educational and regulatory approaches (discussed in Chaps. 7 and 8) have begun to show a temporal association at least with reduction in opioid prescribing and possibly in some morbidity and mortality outcome measures as well. There has in fact been a shift noticed by emergency department physicians and law enforcement within the past couple years toward “street” opioids such as heroin, as the procurement of prescription drugs obtained illicitly is becoming more difficult. (While it is within the realm of responsibility of the physician to educate and treat patients who abuse street drugs, controlling the transmission of such remains the responsibility of law enforcement and as such is not discussed in this book.) At any rate, the often-quoted analogy of a pendulum swinging back and forth between poles of liberal vs. conservative prescription of opioids for pain seems to be traversing back toward the guarded at present. While this undoubtedly represents a public health triumph in many regards, with reductions in morbidity and mortality, it must not be forgotten that in the majority of cases, opioid-dependent individuals suffer from various predisposing chronic pain states.
In the United States, the prevalence of chronic pain has been estimated at 100 million individuals [1]. This of course ranges from aggravating single-limb osteoarthritic pain and dysfunction to global, entire-body states of agony. Associated disability which also varies tremendously among persons with the same apparent pathology further adds to the misery of the individual and often the family and in some cases society as well. As the preface to the Institute of Medicine’s (IOM) landmark 2011 monograph, “Relieving Pain in America ” states:
To underscore the seriousness of the issue, if empathy for suffering is not enough, it should be noted by all practitioners of every healthcare discipline that suicide is two to three times more prevalent in the chronic pain population than the general population [2, 3].
The magnitude of the pain suffered by individuals and the associated costs constitute a crisis for America, both human and economic…. [1]
As physicians, we are ethically and professionally responsible to alleviate pain and suffering whenever possible. Understanding the multifaceted nature of both pain and suffering (biopsychosocial-spiritual) is essential to accurate assessment and thus effective treatment. Unfortunately, the training of most physicians and mid-level providers is sadly lacking in at least one of these dimensions, and these deficits have been highlighted recently as a major issue requiring educational and even certification-oriented overhaul [4]. To quote the IOM report again:
The recently unveiled National Pain Strategy [4] emphasizes as one of its core foci the remediation of what comprises a substantial deficit in provider knowledge and competency of how pain works and how to treat it.
Many health care providers lack a comprehensive perspective on pain… We believe pain arises in the nervous system but represents a complex and evolving interplay of biological, behavioral, environmental, and societal factors that go beyond simple explanation. Knowledge of pain needs to be enriched from the molecular and genetic to the cellular, neural network, and systems levels. It is necessary to understand how the settings and surroundings in which pain occurs and is experienced have an impact on its biology. [1]
This chapter is divided into two main components: first is a basic primer on pain physiology and pathophysiology and psychology . It is certainly not intended to serve as an exhaustive treatise on the subject; for more complete sources, the reader is referred to the excellent textbooks by Wall and Melzack [5], Bonica [6], and Deer [7], among others. Physical pain has classically been held to be a cardinal symptom of inflammation and many other disease processes; while many champion the notion that chronic pain is a disease in its own right [1], it remains evident that pain is always a symptom. As such, any complaint of pain requires thoughtful history taking and physical examination (both of which in themselves are often therapeutic for patients whose complaints have not been addressed to their satisfaction), frequently the formulation of a differential diagnosis, and then frank discussion about natural history and therapeutic options.
The second half of the chapter focuses on multimodal pain therapy —alternatives to opioids. Prevention (discussed in greater detail in Chap. 11) must undergird every effort of the physician to help the patient complaining of pain. Poor dietary choices, tobacco and alcohol use, sleep deprivation, sedentary lifestyle, and many other physical insults are in many cases contributory if not causative of a great many pain states. Psychological dysfunctions (including anxiety and stress, cognitive distortions, and deeper underlying emotional wounds) often amplify—if not initiate—chronic pain. A proper viewpoint on the spirituality of the individual and how that spirituality relates to their pain experience is essential to treating the whole person. Concurrent with efforts aimed addressing these issues, rational treatment must apply (or at least offer) interventions that maximize the benefit-risk ratio not only for the individual intervention in question but for the entire treatment plan, whether or not opioids are part of that plan.
Pain Basic Science Primer
Before briefly discussing common therapeutic modalities for treating pain, it is important to take a step back and examine what we think we know about pain. Everyone knows what pain is, but no one can fully explain it. Definitions are generally circular (“It hurts!”). The following International Association for the Study of Pain (IASP) definition of pain seems excessively vague at first glance but, as we will see shortly, is of necessity very nonspecific:
An unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage. [8]
Nociception and the Cartesian Model
As with most scientific ideas, the conceptualization of what pain is has undergone numerous paradigm revolutions since the Renaissance. In 1664 Rene Descartes proposed that pain may have more to do with physical stimulus and cerebral perception than prior constructs based on metaphysical, spiritual/moral, or other ethereal factors (Fig. 6.1). The basic assumption that insult to the physical integrity of the organism is transmitted and translated into the subjective experience of pain is so entrenched in our thinking 450 years later that it is difficult for most to conceive of a model of pain that differs much from this. Furthermore, laboratory and clinical investigation have verified and elaborated on the basic injury stimulus-pain response pattern to the point that it is very nearly dogma that if pain is present, i.e., “organic” pathophysiology, tissue damage is responsible for it.
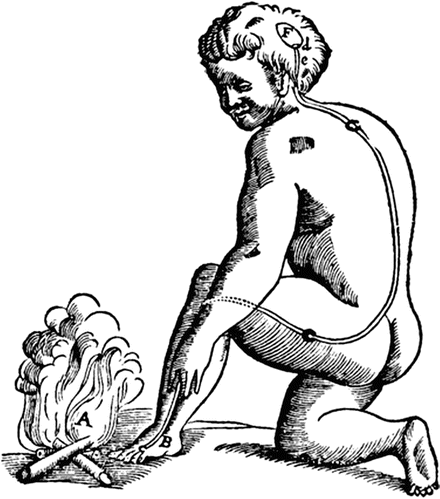
Fig. 6.1
“For example, if the fire A is close to the foot B, the small particles of fire, which as you know move very swiftly, are able to move as well the part of the skin which they touch on the foot. In this way, by pulling at the little thread cc, which you see attached there, they at the same instant open e, which is the entry for the pore d, which is where this small thread terminates; just as, by pulling one end of a cord, you ring a bell which hangs at the other end… Now when the entry of the pore, or the little tube, de, has thus been opened, the animal spirits flow into it from the cavity F, and through it they are carried partly into the muscles which serve to pull the foot back from the fire, partly into those which serve to turn the eyes and the head to look at it, and partly into those which serve to move the hands forward and to turn the whole body for its defense.” Descartes’ Traité de l’homme, 1664. US Public Domain
This simplest of frameworks, cause and effect, of course has quite a bit of truth to it. In the conscious patient, nociception (the activation of and communication from peripheral pain receptors on specialized nerves described in more detail below) is a real and in many cases sufficient cause for the experience of pain, especially acute pain. (The criteria of consciousness applied in the previous sentence are based upon the currently accepted definition of pain as a conscious subjective experience.) Evidence supporting the importance of nociception is consistently found in the operating room when the administration of intravenous opioids or ketamine or institution of conductive nerve block to a sedated/anesthetized patient results in normalization of vital signs and suppression of EEG activity and unconscious aversive behavior. Not to mention, almost everyone has slammed their finger in a car door or stubbed their toe and experienced the (subjective) effect.
Understanding pain should begin with understanding nociception. Somatic nociception, which arises from non-visceral sources, is discussed first; visceral nociception is functionally very similar but more complicated due to richer autonomic communication. Nociceptors are specialized receptors on sensory neurons among the Aδ- and C-fiber families (discussed in greater detail below) that generate an afferent action potential in response to a specific noxious stimulus. This process of transduction occurs in response to mechanical, thermal, or chemical stimuli generally associated with tissue damage and is initiated by transmembrane cationic channels such as transient receptor potential (TRP) and tetrodotoxin-resistant or tetrodotoxin-sensitive sodium channels. Besides activation by initial stimulus, nociceptors may be sensitized (with lowered thresholds or in some cases even “awakening” of “silent” nociceptors) by subsequent local inflammatory mediators including arachidonic acid, bradykinin, histamine, serotonin, substance P, calcitonin gene-related peptide (CGRP), nerve growth factor, potassium, hydrogen ions, and other factors. This peripheral sensitization may result in the phenomenon of hyperalgesia (pain sensed with less-than-normal stimulus intensity) and allodynia (painful perception of a typically non-painful stimulus such as light touch). Besides pain (dolor), these mediators elicit other cardinal inflammatory symptoms and signs including vasodilatation and extravasation leading to heat (calor), erythema (rubror), and edema (tumor).
These first-order neurons, with cell bodies located in the paraspinal dorsal root ganglia and long axonal projections to the periphery, are classified as Aδ– and C–fiber neurons, with distinct roles. Larger, myelinated Aδ fibers (also known as “fast pain fibers”) conduct relatively rapidly and provide fairly precise pain localization due to a small receptive field. Sharp, stabbing, acute sensations comprise the message carried by the fast pain fibers. In contrast, unmyelinated C fibers (“slow pain fibers”) which outnumber their Aδ counterparts 2:1 conduct much more slowly and confer less precise localization of pain. Slow pain fibers conduct aching, sore, and burning pain. Both sets of afferents enter the dorsal horn of the spinal cord (along with other sensory traffic) either directly or after a one-to-two segment rostrocaudal deviation through Lissauer’s tract and synapse with their second-order neuron (also known as projection neurons) in specific gray matter regions delineated as the Rexed laminae .
Aδ fibers synapse primarily in lamina I (also laminae II, V, and X) and communicate with their projection neurons primarily by means of glutamate. Aspartate is another excitatory neurotransmitter involved in communication between first-order and projection neurons, and multiple other facilitatory (e.g., substance P, CGRP) or inhibitory (e.g., gamma-aminobutyric acid) neurotransmitters are involved in enhancing or suppressing transmission, as discussed in greater detail below. Projection neurons synapsing with the Aδ fibers generally decussate at the level of cord entry and ascend via the spinothalamic/anterolateral tract to the thalamus (ventroposterolateral nucleus or VPL). Similarly, primary Aδ afferents from the head and face relay their messages via the trigeminal system, descending in the brainstem to the medullary spinal trigeminal nucleus (nucleus caudalis), synapsing with a second-order neuron that also decussates and joins the spinothalamic tract to ascend to the thalamus (ventroposterior medial nucleus or VPM). From the thalamus, these fast pain pathways project via third-order neurons to the somatosensory cortex and are represented along with other sensory traffic somatotopically on the postcentral gyrus (Fig. 6.2).
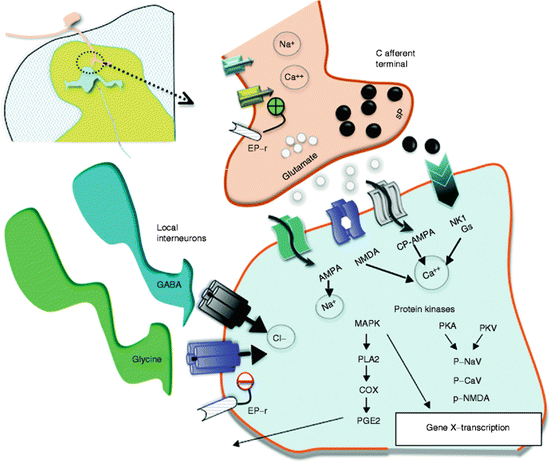
Fig. 6.2
This schematic provides an overview of the organization of the events transpiring at the level of the first-order synapse. (1) As indicated, the presynaptic effects of depolarization lead to opening of voltage-sensitive calcium and sodium channels with increases in intracellular sodium and calcium and mobilization and release of transmitters (sP and glutamate). (2) These act upon eponymous receptors (see text), leading to depolarization and increase in intracellular calcium. (3) Activation of kinases which phosphorylate a variety of channels and receptors activates intracellular enzyme cascades such as for PLA2 and increasing gene transcription. (4) Release of products such as prostanoids (PGE2) which can act upon the local membrane through their eponymous receptors (EP-r) where presynaptically they enhance the opening of voltage-sensitive calcium channels and postsynaptically reduce the activity of glycine receptors. (5). As indicated in addition, the first-order synapse is regulated by inhibitor interneurons such as those release GABA and glycine. These interneurons can be activated by afferent collaterals and by descending pathways to downregulate the excitability of this synapse. Reprinted from Comprehensive Treatment of Chronic Pain by Medical, Interventional, and Integrative Approaches, A Survey of Systems Involved in Nociceptive Processing, 2013, Yaksh TL, Wiese AJ. With permission of Springer
C fibers are a more functionally heterogeneous group than their Aδ counterparts, with a greater diversity of both activating stimuli and output message (see Todd and Koerber’s excellent summary of C-fiber biology [9]). They generally synapse in lamina II, also known as the substantia gelatinosa (and to a lesser extent in laminae I and V) and communicate primarily with the interneurons that make up the primary population of that lamina, via both substance P and glutamate. Many of their ultimate second-order afferents (especially those in lamina V) are often described as “polymodal” or “wide dynamic range” neurons, indicating their ability to receive sensory messages from multiple input sources besides nociceptors. Some of these projection neurons then decussate (while others do not) and both ascend via the paleospinothalamic tract in the anterolateral region of the spinal cord to the reticular formation of the brainstem and to the centromedian and parafascicular intralaminar thalamic nuclei, from whence third-order neurons project to a more diverse cortical audience including insula, cingulate, and frontal cortices.
Visceral nociception is far less understood. Visceral nociceptors respond a much more limited array of stimuli than their somatic counterparts; for example, it is well known that most mechanical insults to the gut including cutting are not perceived. Visceral sensation is relatively limited in physiologic states to detecting distension; in pathology however inflammation and ischemia produce pain. Abdominopelvic organs are innervated by multiple networks including parasympathetic and sympathetic efferents and general visceral afferent (C, Aδ, and Aβ) fibers carrying polymodal sensory information. The alimentary canal also contains a dedicated neural network known as the enteric nervous system , which besides controlling motility and absorptive and immune functions also modulates local inflammation and can sensitize the visceral afferents via mediators including serotonin, substance P, and CGRP [10]. Visceral afferents project via both sympathetic and parasympathetic nerves, with the former held to be the primary pathway for visceral nociception. These sympathetically associated Aδ and C fibers run a tortuous course from the organ to the dorsal horn, primarily within the greater splanchnic, lumbar colonic, and hypogastric nerves, and along with (but in reverse order to) their sympathetic counterparts traverse the prevertebral (celiac, superior, and inferior mesenteric) ganglia and paravertebral ganglia and enter the dorsal ramus via rami communicantes to their cell bodies located in the dorsal root ganglia. From there, the pathway is analogous to that of somatic afferents, entering the dorsal horn and synapsing with a second-order neuron generally in Rexed laminae I, V, or X [11, 12]. The second-order neuron then ascends to the thalamus, in particular the posterolateral nucleus [10]. Visceral afferents also accompany parasympathetic (vagal and pelvic) nerves; these appear to be relatively more important than their sympathetic-associated counterparts in mediating sensory (including nociceptive) information from lower structures including the colorectum, urinary bladder, and genitalia [10]. Finally, recent human and animal evidence also point to a significant dorsal column visceral nociceptive pathway [10] that may be even more important in mediating visceral pain than are spinothalamic or spinoreticular tracts. This pathway has no known somatic correlate, as the dorsal columns mediate only somatic tactile and proprioceptive sense, to our current knowledge.
The distinction between “somatic” vs. “visceral” pain based is especially clinically relevant when evaluating pain in the thorax and abdomen. Abdominal pain, for example, can arise from either internal organs (generally poorly localized and more frequently associated with autonomic phenomena) or from somatic sources such as the abdominal wall or an intercostal nerve (more clearly delineated). The concept of referred pain should be addressed briefly as well, which is a phenomenon whereby pain arising from pathology in one region is perceived in a somatically distinct (and sometimes distant) region. Referred pain is most commonly considered as a visceral phenomenon (e.g., right scapular pain with cholecystitis, jaw claudication, and left arm pain with myocardial ischemia); however, a wealth of data and experience indicate that somatic pain generators such as zygapophyseal joints [13] and myofascial trigger points [14] also refer pain elsewhere. Visceral pain referral is thought to occur as the result of convergent or shared pathways between visceral and somatic afferents at the dorsal horn, with some evidence for brain-level convergence as well [15].
While in no way intended to disparage the genius of Descartes and his groundbreaking theories at the time, the caption in Fig. 6.1 is also reproduced from his classic treatise to illustrate the possibility that 400 some years from now, people may look back on our current theories and models as equally quaint.
Neuropathic Pain and Central Sensitization
Neuropathic pain is currently defined by the IASP as “Pain caused by a lesion or disease of the somatosensory nervous system…Neuropathic pain is a clinical description (and not a diagnosis) which requires a demonstrable lesion or a disease that satisfies established neurological diagnostic criteria” [8]. The term “non-nociceptive pain” is often errantly used as a synonym for neuropathic pain; this imprecise nomenclative admixture is discouraged, as much non-nociceptive pain falls outside the realm of objective neurologic pathology.
Neuropathic pain is often characterized by spontaneous and often lancinating pain and multiple other fairly consistent symptoms (e.g., burning sensation, dysesthesias) and signs (e.g., hyperalgesia and allodynia, temporal summation). Neuropathic pain encompasses both well-localized/anatomically defined syndromes such as carpal tunnel syndrome, sciatica, and tic douloureux and more widespread (typically distal extremity) burning pain seen in advanced diabetes, hypothyroidism, vitamin B12 deficiency and with neurotoxicity from alcohol, heavy metals, or chemotherapeutic agents. CNS lesions such as spinal cord or thalamic/cortical lesions may confer even more generalized and devastating pain states. Proposed mechanisms underlying the genesis and perpetuation of neuropathic pain include [16–19]:
Persistent inflammatory state at the site of the lesion or upstream sites (e.g., dorsal root ganglion) may sensitize/lower the nociceptive threshold of the primary afferent.
Increased TRP and sodium channel expression on primary afferents, with decreased action potential threshold/increased excitability, and frequently ectopic potential generation.
Increased adrenoreceptors (α1 and α2) with increased sympathetic sensitivity.
Activation of N-methyl-D-aspartate (NMDA) receptors in the dorsal horn leading to “windup ” (increasing sensitivity to nonincreasing stimulus intensity) and lowered threshold/tonic activation of secondary afferents.
Loss of descending inhibition (discussed in greater detail below).
“Invasion” of Aβ afferents into the Rexed lamina II with normally non-nociceptive primary afferent stimuli now evoking pain (allodynia).
Recruitment of typically non-nociceptive secondary afferents into a nociceptive state.
Microglial activation/sensitization with resultant CNS pro-inflammatory effects.
Inherent in this construct is the apparent active and dynamic role that both peripheral and central nervous systems play not only in communicating and interpreting pain but also in amplifying and even generating it. No longer viewed as merely a passive relay from the periphery to the brain, nocineurons (and microglia) are now being seen as effectors and modulators of many if not most chronic pain states. While not included in this primer for the sake of brevity, our growing understanding of the critical role microglia play in the chronification or control of pain is deserving of much greater attention, and the reader is referred to the excellent review by Beggs and Salter [20] and others for an introduction to the topic.
Taking the concept of CNS participation in the pain experience a step further, the phenomenon of central sensitization first described by Woolf in 1983 [21] has greatly expanded our understanding of chronic pain. Nociception (and neuropathic pain) is not the full story by any means; non-psychogenic pain can occur in the absence of nociception (or neuropathic pain per IASP definition). Central sensitization provides a plausible explanation for the subjective phenomena of many pain states disproportionate to (even occurring in the absence of) any discernible pathology, along with related features of allodynia. Objective evidence for central sensitization states include changes in functional MRI signal and electrophysiologic parameters (potential amplitudes) [22]. Essentially the theory (with ample animal and human evidence) posits that a reduction in pain threshold has occurred within the CNS such that previously insufficiently noxious or non-noxious stimuli are now painful (hyperalgesia and allodynia, respectively). In Dr. Woolf’s words, “The net effect of central sensitization is to recruit previously subthreshold synaptic inputs to nociceptive neurons, generating an increased or augmented action potential output: a state of facilitation, potentiation, augmentation, or amplification” [23]. Proposed mechanisms underlying this state are similar to those presented above for neuropathic pain and include [22, 23]:
Reduced secondary afferent thresholds
Reduced inhibition
Expansion of secondary nocineuron receptive field
Transformation of secondary nocineurons to wide dynamic range neurons capable of activation by formerly innocuous stimuli
Glial contributions
Glutamate (via NMDA and other receptors) is currently thought to be the most important protagonist in the development of central sensitization, effecting these pathologic changes via the establishment of supranormal intracellular calcium levels [23]. However, many other familiar factors such as substance P, CGRP, brain-derived neurotrophic factor, bradykinin, and nitric oxide are invoked in facilitating these changes, which may occur in numerous brain structures as well and are not limited to the dorsal horn [23].
Many chronic pain conditions, including migraines, TMJ disorders, fibromyalgia, osteoarthritic and chronic low back pain, interstitial cystitis, and many neuropathic pain states including complex regional pain syndrome (formerly known as reflex sympathetic dystrophy or causalgia) are now thought to represent manifestations of a central sensitized state. Functional abdominal pain, chronic pelvic pain, and other visceral pain states are also now considered to reflect a centrally sensitized state [24].
Cancer Pain
Cancer is frequently associated with severe chronic pain; it is estimated that over 2/3 of patients with advanced cancer suffer from significant pain related to the disease [25]. Multiple pathogenic (and iatrogenic) processes are responsible for the pain associated with cancer, including [26]:
Local pressure and destruction from the primary tumor
Release of several inflammatory mediators (including bradykinin, nerve growth factor, proteases, tumor necrosis factor α, various interleukins, and cytokines)
Peripheral neuronal injury and alterations of the dorsal root ganglion and neo-neurogenesis and neuroma formation with upregulated sodium channels and ectopic activity
Epidural compression of spinal nerve roots from tumor mass effect (this occurs in up to 10% of cancer patients, and persistent or dramatic back pain complaints require thorough investigation and workup)
Central sensitization with upregulated NMDA and AMPA activity
Osteoclastic activity at bone sites, facilitated by the acidic environment generated by cancer cells
Neuropathy from chemotherapeutic agents (especially vinca alkaloids, the taxanes, and platinum-based compounds)
Neural Plasticity and Chronic Pain
Much recent research has focused on the contribution of neural plasticity to chronic pain, with the former defined by the National Institutes of Health’s 2009 Blueprint for Neuroscience Research Workshop as:
While these changes can occur at any level of the CNS including the dorsal horn as introduced above, that which occurs in the brain is the focus of this section. Distortion of size and other geographic perceptions of injured body parts have been documented for centuries, with phantom limb pain perhaps being the most salient example. Reorganization of the primary somatosensory cortex in response to both acute and chronic pain has been shown over the past two decades to underlie these phenomena [28–31], with the recent application of noninvasive imaging modalities demonstrating functional [32–34] and even anatomic changes [35–38] within the brain in various chronic pain states. Fortunately, it has been demonstrated consistently that these distortions and cortical reorganizations can be undone with adequate therapy [39]. A recurring observation with chronification of pain is that of enhanced and in many cases spontaneous activity in the cortical-limbic circuitry [40–42] and in particular the anterior cingulate cortex , which is associated with a significant emotional component [43]; this has led many to conclude that much if not most chronic pain represents a learned state or:
The ability of the nervous system to respond to intrinsic or extrinsic stimuli by reorganizing its structure, function and connections. [27]
Persistence of the memory of pain and/or the inability to extinguish the memory of pain evoked by an initial inciting injury. [41]
Perception and Modulation
Arithmetic is necessary in constructing a ledger or a house; however, arithmetic, calculus, astrodynamics, and even general relativity are also required to build a vessel and send it to the Moon or Mars and back. The simple Cartesian model may be sufficient to explain acute nociceptive or even neuropathic pain, but falls quite short of explaining the complexities of most chronic pain and suffering, or why such immense variability in subjective experience exists between people afflicted by identical pathology. Not only can pain occur without nociception (or neuropathic pain); nociception can also occur without pain. In the conscious individual, it is quite clear that intangible and immeasurable factors can override or sufficiently modify the simple physical stimulus-response model of pain, as evidenced by Asian fire walkers, Native American Sun dancers, and elite athletes of all disciplines and regions (Fig. 6.3).

Fig. 6.3
Ear pull at the 2007 World Eskimo Indian Olympics, held in Anchorage, Alaska. Reprinted with permission from Patrick Endres, AlaskaPhotoGraphics
Gate Control Theory
Widely regarded as the most seminal work of the twentieth century in understanding pain, Melzack and Wall’s landmark 1965 publication [44] described a “gate control system ” exerting dynamic and variable influence on synaptic transmission between first- and second-order pain afferents. What have come to be known as interneurons were postulated as assessing and responding to competing inputs from both peripheral non-nociceptive afferents as well as from brain regions to allow or prohibit nociceptive stimuli from proceeding rostrally. The gate control theory expanded our understanding of the role of the central nervous system in processing and even influencing pain. In Dr. Melzack’s words, it:
Prior to our (yet superficial) understanding of the neurobiology of pain, practitioners across the world for millennia have used various means of modifying pain perception with sometimes astounding success (witness the reports of efficacy of mesmerism or acupuncture in facilitating operations without allopathic anesthesia). Dr. Henry Beecher (introduced in Chap. 2) was the first in the modern era of medicine to document and report on the potency of psychological factors in modifying pain. He observed that soldiers sustaining severe wartime injuries were often apparently unmoved by their trauma, exhibiting little pain behavior in the acute and subacute phases of their injury [46]. This was noted to be in marked contradistinction to similar injuries sustained by civilians outside of the war zone and also in sharp contrast to pain behaviors later demonstrated by these same soldiers in response to minor nociceptive stimuli such as venipuncture. He surmised that the profound emotional influences of surviving potentially lethal combat, the knowledge that they would be returning home, and the camaraderie of their fellows, among other factors all contributed to suppression of pain perception [47]. We now understand that these soldiers’ stoic responses to what would otherwise typically elicit significant pain response were a manifestation of descending modulation . (As a side note, it is fascinating that Dr. Beecher’s most important legacy is the placebo-controlled trial, as we now understand that the factors involved in mediating the placebo response are those involved in the process of descending modulation.) By the 1950s, it was evident that descending projections from the brain could modulate afferent traffic [48, 49], and by the late 1960s, it had been demonstrated that focal electrical stimulation of the midbrain produced sufficient analgesia to allow for highly noxious surgical procedures to transpire in the absence of any other anesthetic [50]. This (invasive and experimental) phenomenon has been replicated in humans, allowing preservation of general cognitive as well as motor function while eliminating the perception of pain [51, 52]. Intensive investigation over the subsequent half century has revealed a complex bidirectional (descending and ascending) modulating network that functions to suppress or enhance pain transmission according to a host of competing inputs and the needs of the organism [53, 54]. Teleologically, it makes sense that pain, despite being a vital “sense,” must be triaged along with other inputs such as hunger or thirst or the need for sleep. Accordingly, the system may function to augment the pain message when amplification is required for benefit or to dampen the pain message when competing needs (e.g., running from a predator) supersede. Pathologically, the amplification of the system may be seen in many chronic pain states as introduced in the previous section on neuroplastic pain and sensitization. Conversely, enhancement (or simple utilization) of the system’s ability to suppress pain may be harnessed therapeutically, as practitioners of spirituality, meditation, exercise, and various healing arts have done throughout recorded history.
Forced the medical and biological sciences to accept the brain as an active system that filters, selects and modulates inputs. The dorsal horns, too, were not merely passive transmission stations but sites at which dynamic activities (inhibition, excitation and modulation) occurred. [45]
Related posts:
 Best Practices Education, Part III: Regulatory and Advisory Issues Related to Opioid Therapy for Pain
Best Practices Education, Part III: Regulatory and Advisory Issues Related to Opioid Therapy for Pain
 Best Practices Education, Part II: Evidence for and Against Opioid Therapy
Best Practices Education, Part II: Evidence for and Against Opioid Therapy
 Attenuating the Agent: Reducing Opioid “Virulence”
Attenuating the Agent: Reducing Opioid “Virulence”
 Addressing Host Factors: Primary, Secondary, and Tertiary Prevention of Opioid Dependence
Addressing Host Factors: Primary, Secondary, and Tertiary Prevention of Opioid Dependence
 Opioid Dependence Risk Factors and Risk Assessment
Opioid Dependence Risk Factors and Risk Assessment
 Addressing Host Factors: Overview of Dependence and Addiction
Addressing Host Factors: Overview of Dependence and Addiction
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


