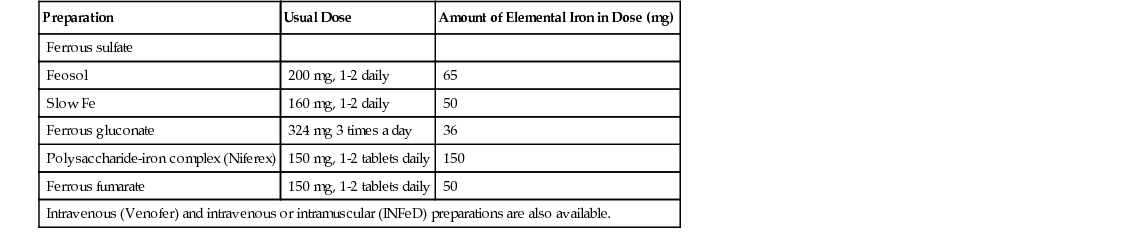
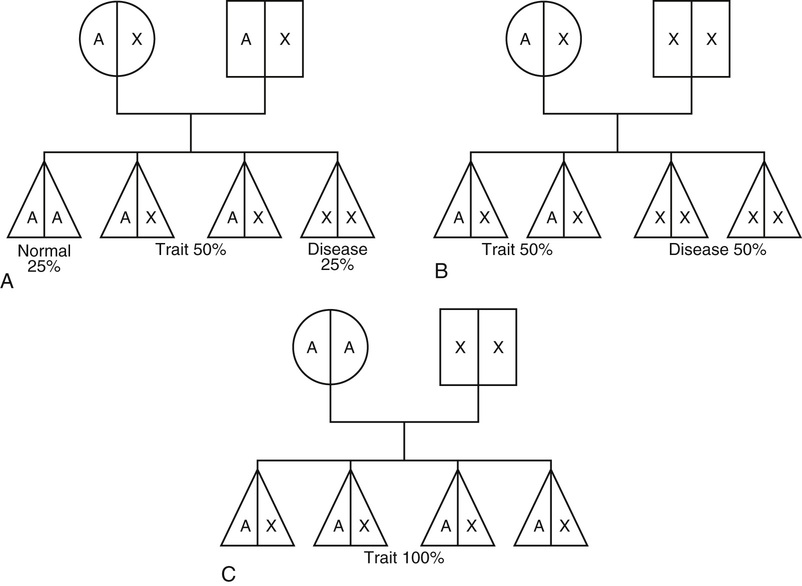
Margaret M. Sennett Anemia is not a disease but rather a sign or symptom of an underlying disorder. Anemia is defined as a reduction in the number of red blood cells (RBCs), hemoglobin concentration, or hematocrit. In general, a hemoglobin concentration in adults below 13.6 g/dL for men or below 12 g/dL for women suggests anemia.1 Although certain signs and symptoms are sometimes associated with anemia, the diagnosis is often based on laboratory data alone. As one of the most common hematologic diagnoses, anemia affects people of every age group. Some types of anemia have a higher incidence in certain races and ethnicities. For example, sickle cell disease is most common in those of African ancestry, and thalassemia in people from the geographic regions of the Mediterranean, the Middle East, Southeast Asia, and parts of India and Pakistan. Anemia is a condition in which too little oxygen is being transported to the tissues. Inadequate supplies of oxygen in the tissues produce the symptoms of anemia. There are many types of anemia with a variety of different causes. Grouping them into the categories of (1) RBC production disorders, (2) RBC destruction disorders, and (3) anemia from blood loss enables a clear understanding of the pathophysiology of this disease. Erythropoiesis, the production of erythrocytes (RBCs), is limited to the axial skeleton and proximal ends of the long bones in the adult. The marrow is a special environment for hematopoietic development and consists of a matrix of reticular cells and fibers called the marrow stroma. It is here that stem cells are stimulated by colony-stimulating factors to grow and differentiate into the various blood cells including RBCs.2 The most common disorder related to inadequate RBC production is iron deficiency anemia (IDA; see later). Iron is critical for both erythrocyte proliferation and maturation and for hemoglobin synthesis. Under normal conditions, the turnover rate for erythrocytes is approximately 1% per day and erythrocytes survive 120 ± 20 days. Although, in response to hypoxia and anemia, marrow precursors may be seen in 2 to 3 days, an increase in the reticulocyte count will take several more days.3 The most important factor in the body’s ability to increase RBC production is iron. Without adequate iron stores, the marrow cannot increase erythropoiesis.4 Anemia of chronic kidney disease (ACKD) is also a result of inadequate RBC production. Erythropoietin is a glycoprotein hormone secreted by the kidney that induces erythroid precursor cells to differentiate, thereby increasing new RBC production. The control of erythropoiesis is dominated by the decreased concentration of hemoglobin in the blood. This leads to changes in tissue oxygen tension and in turn triggers receptors within the kidney to secrete erythropoietin. Chronic kidney disease can result in an undersecretion of erythropoietin, thereby suppressing an essential signal triggering RBC production.4,5 Other types of anemia caused by defects in RBC production include vitamin B12 and folate deficiency, anemia of chronic disease (ACD), and aplastic anemia, which is a result of bone marrow stem cell failure.5 The mechanisms involved in the increased hemolysis or destruction of RBCs resulting in anemia include various hemoglobin disorders such as sickle cell anemia and thalassemia; RBC membrane defects as in hereditary spherocytosis and hereditary elliptocytosis; RBC enzyme defects as in glucose-6-phosphate dehydrogenase (G6PD) deficiency; and finally, autoimmune antibody production as seen in autoimmune hemolytic anemia. Acute blood loss resulting from situations such as trauma or severe menorrhagia may result in life-threatening anemia with significant symptoms of hemodynamic cardiovascular compromise. Anemia symptoms resulting from mild to moderate menorrhagia, chronic microscopic hematuria, or chronic occult gastrointestinal (GI) bleeding can be much more insidious because the body compensates for this type of slowly evolving anemia. The presentation of anemia can be variable, depending on the acuteness of onset and the ability of the cardiopulmonary system to compensate. If the patient is healthy and the onset of anemia is gradual, there are few signs or symptoms until the hemoglobin value falls below 7.5 g/dL.6 Patients may initially experience fatigue, malaise, headache, dyspnea, irritability, and a mild decrease in exercise tolerance. Further declines in hemoglobin concentration may be associated with a markedly reduced exercise capacity, resting tachycardia, and dyspnea requiring supplemental oxygen. Other nonspecific findings that can accompany long-term, moderate to severe anemia include wide pulse pressure, midsystolic or pansystolic murmur, confusion, lethargy, brittle nails, glossitis, angular cheilitis, and spoon-shaped nails.4 Pallor of the mucous membranes, lips, conjunctivae, nail beds, and palmar creases is a common sign of anemia. When palmar creases are as pale as the surrounding skin, the patient usually has a hemoglobin value of less than 7 g/dL.6 A head to toe physical examination is warranted in the evaluation of a patient with anemia. Overall appearance suggestive of decreased stamina and energy could suggest fatigue secondary to anemia, or anemia related to another chronic illness. A thorough cardiopulmonary examination including vital signs is essential. Increased heart or respiratory rate or the presence of a systolic murmur may be related to anemia. Special attention should be paid to characteristics of the integumentary system to evaluate for pallor, nail integrity, and signs of angular cheilitis. Also, symptoms of increased bruising may be a clue to a potential bleeding disorder contributing to blood loss and iron deficiency. Diagnostic evaluation of anemia initially should include a complete blood count (CBC) including platelet count, white cell differential, RBC morphology, reticulocyte count, and a peripheral blood smear. Evaluation of all blood cell lines is essential to confirm a sole anemia diagnosis, as opposed to a primary bone marrow disease such as aplastic anemia or an infiltrative process such as leukemia. Of the multiple indexes noted on a CBC report, the mean corpuscular volume (MCV) is the single most useful. Describing RBCs as microcytic, normocytic, or macrocytic based on the MCV determines what other testing is necessary to determine the cause of the anemia. The reticulocyte count is the most easily accessible method of evaluating bone marrow production of RBCs (a direct bone marrow examination requires an invasive procedure). The reticulocyte count provides an assessment of whether the causative factor of anemia is related to either decreased production or increased loss. A normal reticulocyte count is 0.5% to 1.5% of the total RBCs. A normal absolute reticulocyte count (ARC) is 25,000 to 75,000/µL. Any value higher than 100,000/µL is considered a marrow that is responding normally to anemic conditions. Values below 75,000/µL are considered consistent with impaired (decreased) RBC production.4 However, if reticulocytosis is associated with anemia, hemolysis needs to be ruled out by observing for hyperbilirubinemia (marker of hemoglobin catabolism) and increased serum lactate dehydrogenase (LDH), which is indicative of direct cellular injury. Also, a low serum haptoglobin value is a sign of intravascular hemolysis because haptoglobin binds the free hemoglobin that is released into the circulation as erythrocytes rupture. These hemoglobin-haptoglobin complexes are removed from the circulation by the reticuloendothelial system. Hemolytic anemias are complex to evaluate and diagnose; if they are suspected, referral should be made to a hematologist. A low reticulocyte count with anemia points to impaired erythropoiesis indicating a reduction in RBC precursors or ineffective production. Ineffective production is reported as erythroid hyperplasia on the bone marrow biopsy and aspiration report. This means the RBCs are being produced but are not viable and usually do not leave the marrow. Box 237-1 identifies conditions associated with either reticulocytosis or a decreased reticulocyte count. A peripheral blood smear is critical in the evaluation of anemias. RBC size and shape variations as well as abnormal cell populations too small to change the indexes can be directly visualized. For example, as iron deficiency progresses, microcytic cells are noted on the smear long before the indexes change. Evidence of hemolysis as well as abnormal shaped cells (such as sickle shape or spherocytes) on the smear may offer additional clues as to the cause of the anemia. Hemoglobin electrophoresis allows hemoglobin chains to be separated according to differences in the charges of their subunits. It is essential for accurate diagnosis of thalassemias and hemoglobinopathies. Evaluation of the body’s iron stores includes serum ferritin, serum iron, total iron-binding capacity (TIBC), and transferrin saturation percentage. The serum ferritin level reflects total body iron stores. It is the first laboratory value to become abnormal when iron stores are becoming depleted, even before IDA is reflected in RBC morphology. Ferritin concentrations less than 12 ng/mL indicate absence of iron stores. It is important to remember, however, that ferritin is an acute-phase reactant and may be elevated as a result of inflammation. Inflammation causes the release of tissue ferritins resulting from damage to the liver and other ferritin-rich tissues.7 Serum ferritin levels are low in IDA and normal or elevated in ACD. Serum ferritin is also elevated in conditions unrelated to anemia, such as iron overload (either transfusion dependent or hereditary hemochromatosis), inflammatory disorders, and alcoholism. The serum iron concentration reflects the amount of iron bound to transferrin, a plasma carrier protein that regulates iron transport in the blood. Normal values for serum iron are 65 to 165 µg/dL.7 Transferrin is the transport protein for iron and is measured indirectly by the TIBC. The TIBC indicates the availability of binding sites on the protein for iron transport. Normal values for TIBC are 300 to 360 µg/dL.7 The percentage of transferrin saturation can be calculated from the TIBC and the serum iron values as follows: Normal values for percentage of transferrin saturation are 20% to 50%.7 Anemias are generally divided into three categories based on the size of the RBCs suggesting the underlying condition or disease. RBCs are normally uniform in size and shape, and deviations in their appearance can suggest a specific cause for the anemia. The degree of anisocytosis (variation in RBC size) is determined by looking at the RBC indexes on the CBC and at cell morphology on the peripheral smear. The most useful RBC index is the MCV. The MCV is a direct measurement averaging the RBC sizes in the sample. The RBC distribution width (RDW) is an indirect measurement that indicates the degree of homogeneity of the sample. For example, uniformly small RBCs will have a low MCV and a normal RDW, whereas a sample with mostly small RBCs but some normal RBCs can have a low MCV with an increased RDW, reflecting the heterogeneity of the sample. Based on the MCV, anemias are classified as microcytic (MCV <80 fL), normocytic (MCV 80 to 99 fL), or macrocytic (MCV >100 fL).4 Variations in RBC shape (poikilocytosis) provide important diagnostic clues and in fact are often pathognomonic of underlying disease. Box 237-2 classifies commonly seen hematologic disorders according to RBC morphology. IDA is the most common type of anemia in the world and the most common nutrient deficiency. In the adult population, IDA predominantly affects women of reproductive age and older adults. The most common cause is chronic blood loss, especially GI blood loss or menorrhagia.4 Chronic GI blood losses should be suspected as a cause of IDA in adult men and postmenopausal women. Inadequate nutrition and increased requirements for iron are the principal causes of IDA in children and pregnant women. Box 237-3 lists additional causes of iron deficiency. The worldwide prevalence of anemia in pregnant women is 38%, and more than 50% of that is a result of iron deficiency. Daily prenatal use of iron has demonstrated improvement in maternal hemoglobin concentration and reduction in risk of having low-birth-weight babies.8 Iron is an essential nutrient present in all living cells. The human body contains about 3 to 4 g of iron, with more than 70% contained within hemoglobin. The remainder of the body’s iron is stored in the liver and marrow as ferritin and hemosiderin, and a small amount is bound to transferrin in the blood.3 The normal man has a total body iron content of about 4000 mg. Women of childbearing age have about 2000 mg of total body iron, a significantly lower amount because of menstrual blood loss and lower dietary intake. The average adult normally loses approximately 1 mg of iron each day through the natural process of desquamation of cells from the skin, GI tract, and urinary tract. The adult woman loses an additional 1 mg through normal menstruation. The recommended daily allowance of iron is 15 mg/day in the diet of nonpregnant women and 30 mg/day for pregnant women. Dietary iron is absorbed in the duodenum of the small intestine. The amount of iron absorbed from the intestine is determined by several factors, including the iron content of the meal, the form of iron being ingested, the individual’s iron status, and the presence or absence of other substances that can enhance or inhibit iron absorption (Box 237-4).9 When iron requirements increase or intake declines, the small intestine increases absorption of iron to meet the increased demand. If there is no additional supply of iron to meet this increased demand, the body’s iron stores begin to be depleted. At this point, several hematologic parameters are affected. The ferritin levels decline as body iron stores decrease. As body iron stores are depleted, the transferrin saturation decreases, leading to a reduced supply of iron to the RBC precursors, resulting in impaired (iron-deficient) erythropoiesis. At this stage, however, an overt microcytic anemia may not yet be present. Once the iron stores are truly depleted and no iron is available for erythropoiesis, an overt microcytic, hypochromic anemia is present, which manifests in the CBC by a low hemoglobin concentration. The RBC indexes are the last to change (decreased MCV, mean corpuscular hemoglobin [MCH], and mean corpuscular hemoglobin concentration [MCHC]). The peripheral smear will show hypochromia, microcytosis, mild anisocytosis, and poikilocytosis. Iron studies will show a low ferritin level and high TIBC. Mild to moderate iron-deficient states are not associated with any clinical symptoms. Patients with severe IDA exhibit the same signs and symptoms of any type of severe anemia. Patients may complain of fatigue, decreased exercise tolerance, weakness, palpitations, irritability, and headaches. Complaints that are specifically related to iron store depletion include paresthesias, sore tongue, brittle nails, spoon-shaped nails (koilonychia), and pica for starch, ice, or clay.5 In fact, a craving for ice (pagophagia) is a common symptom of women with IDA for unknown reasons. As the severity of the anemia increases, several physical changes may become evident. The patient may demonstrate a more forceful apical pulse, tachycardia with exertion, and a systolic flow murmur. Patients may also demonstrate pallor of the conjunctiva, mucous membranes, nail beds, and palmar creases. The characteristic spooning of the nails may also be present. In the older adult, signs of congestive heart failure may be present. IDA is commonly discovered incidentally during a routine CBC. Once IDA is diagnosed, the history may reveal factors that would cause iron deficiency, such as a recent hemorrhage, GI bleeding, menorrhagia, multiple pregnancies, or inadequate nutrition. Iron studies reveal a low serum iron level, decreased serum ferritin, increased TIBC, and decreased percentage of transferrin saturation (Box 237-5). Laboratory changes occur gradually as the iron stores are depleted. The earliest laboratory change is a fall in serum ferritin, reflecting depletion of iron stores. This change is followed by a decrease in serum iron and an increase in transferrin, producing a reduction in the percentage of transferrin saturation to less than 15% (this will drop below 10% as the severity progresses) and an associated increase in TIBC. The first change in the CBC is a drop in hemoglobin. Only with increasing severity and duration do the RBCs become microcytic and hypochromic. The underlying cause of the iron deficiency must be identified. Blood loss by GI bleeding or repeated voluntary blood donation should be suspected until proven otherwise. Older adults with suspected IDA should be thoroughly evaluated for GI cancers, GI bleeding on the basis of nonsteroidal anti-inflammatory drug (NSAID) use, and alcohol abuse. Only a few diseases need to be considered in the differential diagnosis of a microcytic hypochromic anemia. The thalassemias typically have a moderate to severe microcytosis with varying degrees of anemia but normal iron studies. ACD presents a more common diagnostic dilemma. With long-standing chronic inflammatory illnesses such as rheumatoid arthritis, the defective iron supply can result in severe microcytic hypochromic anemia. Iron studies, especially the serum ferritin level, usually differentiate among true IDA, ACD, and thalassemia (Table 237-1). Both IDA and ACD are associated with low serum iron levels. The ferritin level is normal or increased in ACD and decreased in IDA. The TIBC is normal or low in ACD and increased in IDA. TABLE 237-1 Laboratory Values in Microcytic Anemias Microcytosis can occur in patients with inherited sideroblastic anemias; however, these anemias are rare and are related to X-linked genes. Acquired sideroblastic anemias (idiopathic, secondary to drug or toxin exposure, myeloproliferative or myelodysplastic disease) can be microcytic but generally are macrocytic. These conditions are caused by defects in heme synthesis leading to accumulation of iron within bone marrow erythroid precursors and resulting in abnormal erythroid maturation. This anemia is characterized by ringed sideroblasts (erythroblasts with one third or more of the nucleus surrounded by ferritin deposits).3 A bone marrow examination is necessary for diagnosis. The anemia tends to be severe (hemoglobin concentration of 6 g/dL) to moderate (hemoglobin concentration of 8 to 10 g/dL). Treatment of sideroblastic anemia consists of chronic transfusions and iron chelation therapy (to prevent or to treat the transfusion-dependent iron overload). Pyridoxine (vitamin B6) therapy may sometimes partially correct the anemia in patients with hereditary sideroblastic anemia, leaving them with a milder anemia that does not require as many chronic transfusions. Treatment of IDA usually begins with an oral iron preparation. The usual adult therapeutic dose is 150 to 200 mg of elemental iron per day in divided doses until anemia is corrected. The pediatric dosing is 3 mg of elemental iron per kilogram, and liquid preparations are available. Administration should be continued empirically for 4 to 6 months or until the serum ferritin level exceeds 50 µg/L and then stopped. Common side effects of iron preparations are nausea, constipation, heartburn, upper GI discomfort, black stools, and diarrhea. Iron absorption is optimum when iron is taken 30 minutes before meals with ascorbic acid. Absorption can be reduced by as much as 40% to 50% if it is taken with meals; however, iron on an empty stomach can cause more side effects, leading to noncompliance with medication. GI upset, the most common side effect, may be avoided by starting with a single pill per day and slowly increasing to the recommended dose. Once an adequate dose of iron is reached, changes in the hematologic markers should be seen in just a few weeks. The hemoglobin level should begin to rise within 1 to 2 weeks. The MCV should correct within 1 to 2 months, reflecting the normalization of the erythrocyte size. Supplementation with oral iron should continue until the anemia is corrected, and until the underlying cause of the deficiency is corrected, or indefinitely if the cause of the deficiency is chronic. If the anemia is severe, the patient has an iron malabsorption problem, or oral iron is not tolerated, replacement should be by parenteral (intramuscular or intravenous) administration of iron. Patients should be referred to a hematologist for intravenous administration of iron because it has traditionally been associated with adverse reactions. There are newer intravenous iron formulations (iron carboxymaltose and ferumoxytol) that appear to have a much safer profile. The novel structures of these preparations have been shown to reduce the risk of free iron reactions and result in lower immunogenicity. As a result, test doses are not necessary and much higher doses can be administered, thereby reducing the number of administrations necessary to replace iron stores.10 Children require adequate iron intake to meet the increased demands of rapid growth. Nutritional iron deficits are especially common in children with excessive milk consumption at the expense of adequate intake of iron rich foods. Teenage girls often become iron deficient as a result of menstrual blood loss, sometimes accompanied by poor diet in this age group. Iron supplementation during pregnancy is almost always required. Pregnancy places a greater demand on iron stores, especially during the last two trimesters. The additional iron is needed to cover the needs of the developing fetus and placenta and to accommodate the increase in erythrocyte mass that normally occurs during the later stages of pregnancy. Many women enter pregnancy with inadequate iron reserves as a result of prepregnancy menstruation. Iron studies can be difficult to interpret in pregnancy because of the hemodilutional effects of the increase in blood volume during pregnancy, as well as the elevation of ferritin as an acute-phase reaction to the known inflammatory effects of pregnancy. Adequate maternal iron stores are essential to adequate brain development in the fetus.11 Older adults with suspected IDA should be thoroughly evaluated for GI cancers, even when their stools are negative for occult blood. Next to chronic disease, iron deficiency is the most common cause of a microcytic anemia in older adults. Untreated IDA is especially worrisome during pregnancy. IDA may be associated with preterm delivery, low birth weight, and learning deficits. Untreated iron deficiency in all age groups can lead to severe anemia and may be associated with fatigue, falls, and cardiovascular compromise. Most patients with IDA are diagnosed and treated by their primary health care providers. Patients who are referred to a hematologist for any of the aforementioned reasons are generally referred back to the primary health care provider once the anemia has been corrected, or at least once an accurate diagnosis has been made and the patient is receiving stable iron replacement therapy. Referral to a hematologist should be considered for the following reasons: nonadherence to or intolerance of oral iron replacement, persistent IDA necessitating parenteral iron therapy, and persistent microcytic anemia despite iron replacement and the exclusion of other conditions. Other referrals may be required as evaluation for the cause of the iron deficiency progresses, such as referral to an internist or gastroenterologist to exclude GI blood loss or referral to an oncologist to treat any malignant neoplasms (either GI or gynecologic). Women of reproductive age may require referral to a gynecologist or a hematologist for evaluation of severe menorrhagia (the hematology referral may be helpful to rule out von Willebrand disease as a cause of the menorrhagia). Healthy patients with IDA do not require hospitalization. Patients who are unable to adequately compensate for severe anemia may require hospitalization for cardiac or respiratory compromise that may occur. Patients should receive education about the use of iron supplements to ensure adequate treatment and an understanding of the prescribed regimen. Maximum absorption of iron occurs if it is ingested 30 minutes before meals. Substances that can enhance or inhibit iron absorption are listed in Box 237-4. Calcium can significantly inhibit iron absorption. Multivitamins with calcium or dairy products should be taken 1 to 2 hours after an iron supplement. Ascorbic acid may enhance absorption of iron; therefore, concurrent ingestion of foods rich in vitamin C, such as orange juice, should be encouraged. Numerous iron supplementation preparations are on the market (Table 237-2), some with combinations of iron plus stool softeners, slow-release iron, or iron plus vitamin C. Patients who are intolerant of one preparation may find another that produces fewer or no side effects. The health care provider should therefore encourage patients to try various preparations before recommending parenteral iron. Nutritional counseling is an important strategy to prevent further episodes of IDA and should include assessment of the patient’s dietary intake. Assessment should also include the quantity and timing of iron ingestion and other substances that can interfere with iron absorption, such as tea, coffee, chocolate, dairy products, alcohol, and high-fiber foods. Strict vegetarians who rely on vegetable sources of iron instead of animal sources should be encouraged to supplement their diets with iron-fortified vitamins or to add iron-fortified foods to their diet. Patients whose IDA is secondary to other conditions should be encouraged to seek appropriate medical care. Thalassemia is not a single disorder but rather a group of inherited blood disorders caused by variant or missing genes that affect how the body makes hemoglobin. The resulting anemia depends on the type of thalassemia inherited and varies from asymptomatic to severe hemolytic anemia. All of the thalassemias, except α-thalassemia of a silent carrier, produce some degree of microcytosis and hypochromia. α-Thalassemia is most commonly found in people with ancestry from Southeast Asia, India, China, or the Philippines. β-Thalassemia is more frequent in those of Mediterranean, Middle Eastern, African, or Asian descent. Beta-thalassemia can be inherited concurrently with genes for the hemoglobinopathies, resulting in conditions such as sickle β-thalassemia (Sβ-thalassemia). Sβ-thalassemia severity is inversely proportional to the amount of β-globin produced. When no β-globin is produced (Sβ0-thalassemia) the condition is almost identical to sickle cell disease.12 Thalassemia affects males and females equally. The manifestations and severity of clinical symptoms depend on the number of chain deletions in the hemoglobin molecule. Normally, adult RBCs contain predominantly hemoglobin A1 (96% to 97% of the cell’s hemoglobin) and only small amounts of hemoglobin A2 (2.5%) and hemoglobin F (<1%).4 Thalassemia abnormalities produce changes in the normal amounts of adult hemoglobin. These quantitative changes are important in the diagnosis of thalassemia. Inheritance of thalassemia occurs in a mendelian-recessive manner12 (Fig. 237-1). The two main types of thalassemia are α (alpha) and β (beta), the two protein chains required to make normal hemoglobin. Four genes (two from each parent on chromosome 16) are involved in making α-globin. If one gene is affected, the person is called a silent carrier and usually has no symptoms. If two genes are affected, individuals are considered carriers and have mild anemia (α-thalassemia trait or α-thalassemia minor). People with hemoglobin H (α-thalassemia intermedia) disease have three genes affected and are moderately to severely anemic.13 When all four genes are affected, the condition is called α-thalassemia major (hemoglobin hydrops fetalis), and most affected fetuses are born prematurely and stillborn or die shortly after birth.13 The patients who survive will require lifelong transfusions and extensive medical care. β-Thalassemia genes are located on chromosome 11; each parent provides one to an offspring. An individual is considered a carrier if one gene is affected; this is known as β-thalassemia trait or minor. When both genes are affected, the condition is either β-thalassemia intermedia, causing moderate anemia, or β-thalassemia major (Cooley anemia) with severe anemia. Differentiation between β-thalassemia intermedia and major depends on the volume and frequency of transfusions.
Anemia
Definition and Epidemiology
Pathophysiology
Red Blood Cell Production Disorders
Red Blood Cell Destruction Disorders
Blood Loss
Clinical Presentation
Physical Examination
Diagnostics
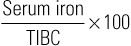
 Physician consultation is recommended for hemoglobin values of less than 10 g/dL in patients with known coronary artery disease and for any patient with postural vital sign changes or active bleeding. Physician consultation is also recommended for sickle cell crises, suspected aplastic anemia, or hemolytic anemia.
Physician consultation is recommended for hemoglobin values of less than 10 g/dL in patients with known coronary artery disease and for any patient with postural vital sign changes or active bleeding. Physician consultation is also recommended for sickle cell crises, suspected aplastic anemia, or hemolytic anemia.
Differential Diagnosis
Microcytic Anemia
Iron Deficiency Anemia
Pathophysiology
Clinical Presentation
Physical Examination
Diagnostics
Differential Diagnosis
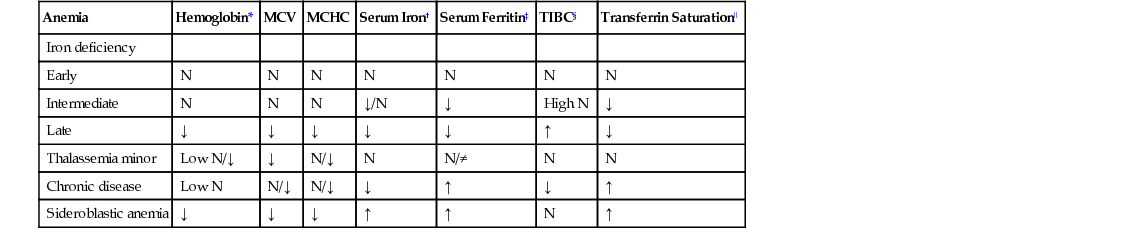
Anemia
Hemoglobin*
MCV
MCHC
Serum Iron†
Serum Ferritin‡
TIBC§
Transferrin Saturation‖
Iron deficiency
Early
N
N
N
N
N
N
N
Intermediate
N
N
N
↓/N
↓
High N
↓
Late
↓
↓
↓
↓
↓
↑
↓
Thalassemia minor
Low N/↓
↓
N/↓
N
N/≠
N
N
Chronic disease
Low N
N/↓
N/↓
↓
↑
↓
↑
Sideroblastic anemia
↓
↓
↓
↑
↑
N
↑
Management
Life Span Considerations
Complications
Indications for Referral or Hospitalization
Patient and Family Education
Health Promotion
Thalassemia
Definition and Epidemiology
Pathophysiology
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Anemia
Chapter 237