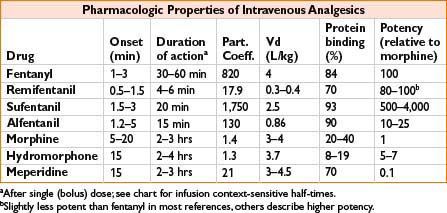
Fentanyl (Sublimaze IV, Fentora Buccal, Actiq Lozenge, Duragesic Transdermal)

Clearance: Liver by CYP-450 3A4; 10% unchanged in urine, metabolite norfentanyl detectable up to 48 hrs
Comments: High lipid solubility causes rapid redistribution to inactive sites (fat, skeletal muscle); therefore, quick onset and quick redistribution below therapeutic index. Duration of action ↑ with large and repeated doses. Peak respiratory depression occurs at 5–15 min (lags behind analgesic effect). Less emetic effect than morphine (see also page 2C-19)
Remifentanil (Ultiva)

Clearance: Unique, rapid metabolism of ester linkage by nonspecific blood and tissue esterases (NOT plasma cholinesterase)
Comments: Bolus produces profound transient analgesia and suppression of autonomic response to noxious stimulus. Rapid recovery observed after infusion regardless of duration (short context-sensitive half-time). May cause bradycardia, hypotension; some data suggest acute opioid tolerance with ↑ postoperative opioid requirements. Do not administer concomitantly with blood as esterases may metabolize. Must dose other analgesics very soon after stopping infusion
Sufentanil (Sufenta)

Clearance: Liver CYP3A4 metabolism, renal/biliary excretion
Comments: Produces hypnosis at doses ≥8 mcg/kg. Calculate dose on ideal body weight. Provides analgesic effect after discontinuation of infusion
Alfentanil (Alfenta)

Clearance: Liver (widely variable)
Comments: Rapid peak effect useful for blunting response to single, brief stimulus (similar to remifentanil). Produced hypnosis as single agent in sufficient concentration. Erythromycin and cimetidine inhibit clearance
Morphine (Astramorph, Duramorph, MS Contin, Others)

Clearance: Primarily renal; metabolites: Morphine-3-glucuronide (55–75%, inactive) and morphine-6-glucuronide, active
Comments: Crosses blood–brain barrier slowly, peak effect may be delayed 10–40 min, complicating titration. Adjust dosing in renal failure. Greatest histamine release of commonly administered opiates. Sustained-release PO preparations available
Hydromorphone (Dilaudid)
Dose: Analgesic dosing: 0.4–2 mg IV (Peds, 0.005–0.02 mg/kg)
PCA dosing: Demand 0.2–0.6 mg q5–15min lockout; basal: 0–0.2 mg/hr (Peds, demand 0.005–0.02 mg/kg q6–20min lockout, basal 0–0.005 mg/kg/hr)
Clearance: Liver metabolism, urine/bile excretion; metabolites: Liver glucuronidation 3-glucuronide (major) and 6-hydroxy (minor)
Comments: Useful alternative to morphine; less histamine release, safer in renal impairment, shorter time to peak effect
Meperidine (Demerol)
Indication: Moderate–severe pain. Also used for postoperative shivering
Dose: Sedation/analgesic dosing: 50–150 mg
IV/IM q3–4h (Peds, 0.5–2 mg/kg IV, IM)
Infusion: 0.3–1.5 mg/kg/hr; postoperative shivering: 12.5–25 mg IV
Clearance: Liver metabolism, urinary excretion
Comments: Direct myocardial depressant, may ↑ HR due to structural similarity to atropine. Metabolite: Normeperidine linked to CNS excitation, seizure caution in elderly, renal impairment, chronic dosing. Administration with MAOI may result in delirium or hyperthermia (serotonin syndrome). Antishivering action may be result of kappa receptor agonism. Shorter duration of action than morphine (see Fig. 2C-1)
Figure 2C-1. Context-sensitive half-time for potent opioids commonly infused during general anesthesia.

Methadone (Dolophine)
Indications: Chronic pain, opioid withdrawal
Dose: Adults, opiate-naïve: Start 2.5–10 mg PO or 2.5–5 mg IV/IM/SC q8–12h; titrate up q3–5d
Mechanism: Opiate agonist and NMDA receptor antagonist
Clearance: Hepatic, renal excretion
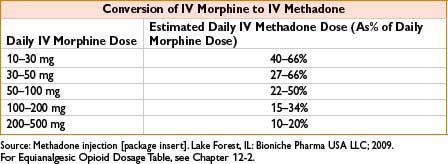
Comments: Respiratory depression is major adverse effect. Very long/variable half-life (13–100 hrs) although many patients require dosing q4–8h to maintain analgesia. Use caution when converting long-standing opiate users to methadone, as paradoxically ↓ dose conversion ratios are required (incomplete cross-tolerance, see the table below). Requires careful titration, deaths reported even in opioid tolerant patients. QT prolongation possible, more common in >200 mg/d; consider EKG when initiating/titrating. Use for detoxification, may require participation of licensed opiate agonist therapy program

Oxycodone
Indications: Moderate–severe pain
Dose: Opioid naïve adults: 5–15 mg (immediate release) PO q4–6h; 10 mg (controlled release) q8–12h, titrate by 25–50% q1–2d
Mechanism: Metabolized to oxymorphone via CYP2D6, poor metabolizers may not achieve adequate effect
Comments: Extended treatment for severe pain to involve scheduled doses of sustained-release, prn availability of immediate-release dose for “breakthrough” pain (similar to other drugs available in immediate/sustained forms). Do not crush or administer sustained-release tablets through feeding tube. Rapid metabolizers (to active form) may have ↑ toxicity. High abuse potential, available in “abuse-deterrent” preparations
Codeine
Indications: Mild–moderate pain, cough suppression
Dose: Adult: 15–60 mg PO q4–6h (max 120 mg/d) Peds: 1–1.5 mg/kg/d divided q4–6h (max 30 mg/d ages 2–6, 60 mg/d ages 6–12)
Mechanism: Metabolized to morphine by cytochrome P450 CYP2D6
Comments: Raises pain threshold without altering response to pain. Lower addictive liability than morphine. Significant genetic variability in metabolism—may cause unreliable analgesia in poor metabolizers or life-threatening intoxication in ultra-metabolizers

Indications: Mild to moderate pain, esp. headaches
Mechanism: Bind to mu receptors with limited response (partial agonist) or no effect (competitive antagonist) and often kappa/delta receptor agonism as well
Comments: Agonist–antagonist can decrease efficacy of subsequently administered opiates. Advantage is that these have limited respiratory depression and ↓ potential for physical dependence. Unlike pure opiate agonists, agonist–antagonists have a ceiling effect in their dose–response relationship (not recommended when pain may ↑). Antagonist effects of these drugs may precipitate withdrawal in opiate-dependent patients. Butorphanol (not nalbuphine) ↑ systolic blood pressure, pulmonary artery blood pressure and cardiac output. Milder effects on GI and biliary systems then with morphine. Butorphanol commonly used in obstetrics; nalbuphine (low dose bolus or infusion) effective in relieving neuraxial opioid-related pruritus without affecting pain control
NMDA RECEPTOR ANTAGONIST & OPIATE AGONISTS
Ketamine (Ketalar)
See also Chapter 2B-8 and Chapter 12
Indications: Used for (1) preincision, intraoperative (“pre-emptive”) analgesia; (2) postoperative opioid adjunct; (3) adjunct in regional and neuraxial anesthesia
Dose*: (1) Bolus of 0.15–0.5 (up to 1) mg/kg and/or infusion at 0.1–0.6 mg/kg/hr; (2) 3 mcg/kg/min × 48 hrs; or 120 mcg/kg/hr × 24 hrs, then 60 mcg/kg/hr × 48 hrs or longer; (3) epidural: 30 mg or 0.25–0.5 mg/kg via epidural before incision. Caudal: 0.5 mg/kg. Intra-articular: 10 mg. Brachial plexus block: 30 mg.
Comments: Potent analgesic with unique mechanism of action. Incremental bolus may be used for short, painful procedures e.g., burn dressing changes. Evidence for decrease in postoperative opioid requirements when low doses used as adjunct. Emerging uses under investigation include sub-anesthetic doses for rescue analgesia in PACU, infusion to improve analgesia in highly opioid tolerant patients, and oral dosing for complex regional pain syndrome as well as neuropathic, ischemic, phantom limb, and cancer pain.
*Studied doses listed, no strong consensus on optimal dose and timing for analgesic adjunct indications.
Tramadol (Ultram)
Dose: Start at 25 mg qAM, increasing 25 mg qd to 25 mg qid, then 50 mg/d × 3 d to 50 mg qid. Max 400 mg/d or 300 mg/d if age >75. May skip dose titration if immediate onset desired
Mechanism: Dual: (1) Opiate agonist (2) spinal inhibition of pain (similar to tricyclics) via inhibition of serotonin/norepinephrine reuptake. Active metabolite M1 has 200× ↑ affinity for mu opioid receptor
Clearance: Hepatic metabolism, renal excretion
Comments: Less respiratory and GI motor effects. May cause seizures, caution in renal disease, alcohol use, stroke, head injury. Potential for life-threatening serotonin syndrome with serotonergic drugs, P450 inhibitors. CYP2D6 poor metabolizers have 20% ↑ tramadol, 40% ↓ M1 levels; genotyping available. Not completely antagonized by naloxone.
OPIATE ANTAGONISTS
Naloxone (Narcan)
Indications: (1) Opiate overdose (severe/life threatening); (2) reversal of opiate respiratory depression; (3) treatment of opiate-induced pruritus
Dose: Adult: (1) 0.2–4 mg IV q2–3min prn (max 10 mg), then may infuse at 0.4 mg/hr and titrate to effect; (2) 0.04 to 0.4 mg doses IV, titrated q2–3min. Infusion: Load 5 mcg/kg, infusion 2.5–160 mcg/kg/hr; (3) 0.25 mcg/kg/hr Peds: (1) Birth to 5 yrs: <20 kg: 0.1 mg/kg IV q2–3min prn; >5 yr or >20 kg: 2 mg/dose q2–3min prn; infusion same as adult. (2) 1–10 mcg/kg IV titrated q2–3min (up to 0.4 mg); (3) 0.25 mcg/kg/hr
Onset: 1–2 min, Duration: 1–4 hrs, depends on route
Mechanism: Competitive inhibition of opioid receptors
Clearance: Hepatic metabolism (95%); primarily renal elimination
Comments: Precipitates opiate withdrawal in opioid-dependent patients; use smallest dose and titrate to desired respiratory rate and level of alertness. May cause hypertension, dysrhythmias, rare pulmonary edema, delirium, reversal of analgesia. “Re-narcotization” may occur because antagonist has short duration, monitor closely and redose as needed. Caution in hepatic failure and chronic cardiac disease
Methylnaltrexone (Relistor)
Indication: Indicated for treatment of opiate-induced constipation and failed laxative therapy
Dose: <38 kg: 0.15 mg/kg SC; 38–62 kg: 8 mg (0.4 mL) SC; 62– 11 4 kg: 12 mg (0.6 mL) SC; typical regimen: Every other day but no more frequently than once/24 hrs
Mechanism: Peripherally acting mu opioid receptor antagonist. Does not cross blood–brain barrier
Clearance: Unknown metabolism. Excreted in urine/feces
Comments: Contraindicated in patients with known or suspected mechanical GI obstruction. In patients with severe renal impairment, ↓ dose by 50%. May cause diarrhea, abdominal pain, nausea, dizziness
TRANSDERMAL/TOPICAL MEDICATIONS
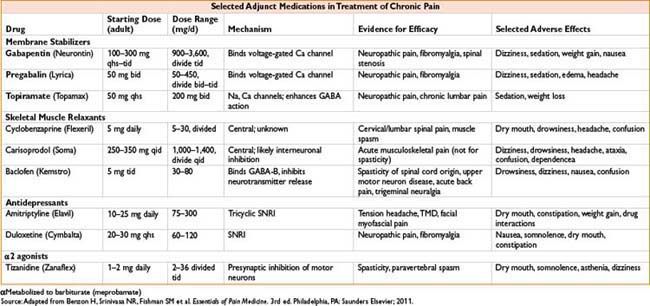
Fentanyl Transdermal (Duragesic)
Indications: Sustained-release opiate therapy, treatment of chronic pain
Dose: See the table below
Mechanism: Opiate agonist
Clearance: See Fentanyl above
Comments: Available from 12.5–100 mcg/hr patches. Time to peak efficacy 12 hrs. Change every 72 hrs. See Fentanyl, 2C-12 for side effects. Conversion from total daily dose of morphine to fentanyl complicated with multiple possible formulas. See the table below. Contraindicated for postoperative pain relief in opiate naïve patients as high risk for respiratory depression. Concurrent use of P450 3A4 inhibitors including some antimicrobials causes ↑ fentanyl levels
Lidocaine Patch, 5% (Lidoderm)
Indications: Neuropathic pain, local inflammatory conditions
Dose: 1–3 patches q24h with 12 hrs on and 12 hrs off typical usage although >3 patches and 24 hrs usage have been studied and found to be safe and well tolerated
Mechanism: Sodium channel blockade
Comments: Produces analgesia but not anesthesia. Minimal systemic absorption. Main side effect is local skin irritation (e.g., burning, dermatitis, pruritus, rash)
NONSTEROIDAL ANTI-INFLAMMATORY DRUGS (NSAIDS)
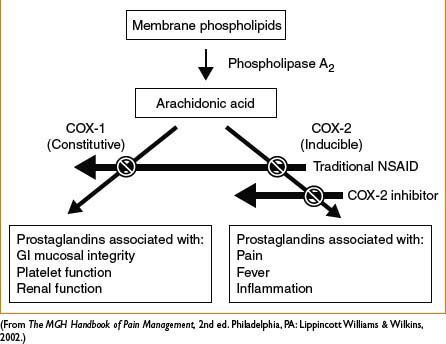
General comments: Produce analgesic, anti-inflammatory, antipyretic effects. Generally have a ceiling effect (unlike opiates) beyond which further analgesia does not occur, but side effects worsen. Mechanism via inhibition of cyclooxygenase (COX) → decreased formation of inflammatory mediators (i.e., prostaglandins). See Figure 2C-2. COX-1 inhibition associated with majority of side effects: GI mucosal ulceration, ↓ renal perfusion, and ↓ platelet aggregation. Inhibition of prostaglandin synthesis suspected mechanism for NSAID-induced bronchospasm. NSAIDs, particularly selective COX-2 inhibitors may ↑ risk of MI and CVA, which can be fatal. Perioperative use in orthopedic surgery requires caution and communication with surgeons; NSAIDs inhibit bone healing in vitro although clinical significance is controversial and under investigation
Figure 2C-2. General mechanism of action of NSAIDs.
NONSELECTIVE COX INHIBITORS
Acetaminophen (Tylenol, Paracetamol, Ofirmev)
Indications: Mild–moderate pain, fever
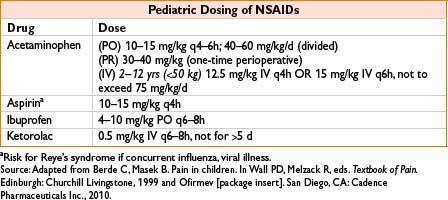
Dose: Adults: PO/PR: 325–650 mg q4–6h, or 1,000 mg q6–8h (max 4 g/d, some recommend max 3 g/d). IV: Adults >50 kg: 650 mg q4h or 1,000 mg q6h over 15 min, max 4g/d; adults <50 kg 12.5 mg/kg q4h or 15 mg/kg q6h max 750 mg/dose or 3.75 mg/d. Peds: See table
Onset: 5 min (IV) 10 min (PO)
Mechanism: Unclear, possibly inhibition of COX-2
Clearance: Liver
Comments: Lacks significant anti-inflammatory effect (not a true NSAID). Favorable side effect profile: Does not produce GI irritation, affect platelet aggregation. Major toxicity in overdose (single dose or cumulative use): Hepatic necrosis due to depletion of antioxidant glutathione, formation of N-acetyl-p-benzoquinone. Acetylcysteine may substitute for glutathione and prevent hepatotoxicity if administered within 8 hrs of ingestion. Some evidence suggests 2–3 g/d may be safe in chronic hepatic disease, use caution. Reduce dose with severe renal disease. Rectal absorption slow and erratic. May be used in pregnancy
Propionic Acid Derivatives: Ibuprofen, Naproxen, Ketoprofen, Diclofenac
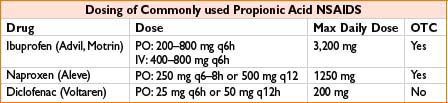
Comments: See general NSAID comments. May exacerbate renal disease, especially in hypovolemia. Extensive protein binding may lead to adverse drug interaction. Ibuprofen has less protein binding than other propionic acids. Note that IV ibuprofen is now available
Ketorolac (Toradol)
Dose: Adult: >50 kg and <65 yrs old: 30 mg IV q6h, max 120 mg/d. ≥65 yrs old or <50 kg: 15mg q6h, max 60mg/d. Max duration 5 d. Peds: 2–16 yrs 0.5 mg/kg IV, then 0.25–0.5 mg/kg/dose q6h up to 48 hrs
Clearance: <50% hepatic metabolism, renal metabolism; 91% renal elimination
Comments: PO formulation available. Parenteral administration makes useful short-term adjunct to parenteral or epidural opioids for severe pain. When administered IV, analgesic > anti-inflammatory effect. Does not cause respiratory depression or biliary tract spasm. Routine doses may be equianalgesic to 10 mg morphine. Effect on platelet function and prolonging bleeding time is observed with spinal anesthesia but not with general anesthesia. Renal injury minimized with adequate hydration. Reduce dose or avoid in elderly and renal insufficiency.
Aspirin, Acetylsalicylic Acid
Dose: Analgesic/antipyretic: 325–650 mg PO q4–6h
Indications: Low intensity pain, headache, musculoskeletal pain, antipyretic
Mechanism: Irreversibly acetylates COX
Comments: Typically stopped 5–10 d prior to surgery due to irreversible antiplatelet effect, which may be desirable for prevention of thrombosis, MI, CVA. Contraindicated in bleeding GI ulcers, hemorrhage, thrombocytopenia, hemophilia. Caution in uremia, von Willebrand’s disease, asthma
SELECTIVE COX-2 INHIBITORS
Celecoxib (Celebrex)
Indications: Osteo- and rheumatoid arthritis, juvenile arthritis, acute pain
Dose: Adult: 200 mg daily, may divide bid. Pediatric: 50 mg bid in patients 10–25 kg
Mechanism: Selective COX-2 inhibition
Comments: Potentially less GI adverse effects. Reduce dose by 50% in moderate hepatic impairment. May also be associated with CV thrombotic events, transaminitis, hypertension, fluid retention, renal injury, allergic or skin reactions (avoid in sulfa, aspirin allergy).
< div class='tao-gold-member'>









