Chapter 31 Acute severe asthma
Acute severe asthma is a medical emergency associated with significant morbidity and mortality. The majority of adverse outcomes are attributed to underestimation of severity with delayed and/or inadequate treatment1–3 and are potentially preventable.
The worldwide prevalence of asthma varies widely (2–37% in children),4 but it is now clear that the overall prevalence has increased worldwide,5,6 with life-threatening episodes affecting an estimated 0.5% of asthmatics per year.7 Australia, New Zealand and the UK have amongst the highest incidences.7 In Australia, 9% have asthma as a long-term condition8 and up to 40% of children have asthma symptoms at some time.5
Although the prevalence of asthma is increasing, many countries have achieved reductions in hospital presentations and admissions,9,10 reduced intensive care admissions and reduced overall asthma mortality.9,10 Improved community management of asthma,1,2 more widespread use of inhaled corticosteroids3 and other preventive measures have been given the credit for these improvements.
Despite reducing admissions, significant and potentially preventable mortality continues to occur in those patients who do require intensive care or mechanical ventilation.11,12
CLINICAL DEFINITION
Asthma has been defined as a lung disease with the following characteristics:13
Exacerbations of asthma are characterised by increasing dyspnoea, cough, wheeze, chest tightness and decreased expiratory airflow. Status asthmaticus has had varying definitions. However, for practical purposes, any patient not responding to initial doses of nebulised bronchodilators should be considered to have status asthmaticus.14
AETIOLOGY
The pathogenesis of asthma is complex, with both genetic and environmental influences. The increase in asthma prevalence has been attributed to the ‘hygiene hypothesis’15 which suggests that reduced exposure to childhood infections as a result of antibiotics and hygienic lifestyle promotes an imbalance in T-cell phenotype leading to inflammatory cytokine overproduction. Immunoglobulin (Ig) E-dependent mechanisms appear to be particularly important in generating the characteristic state of airway inflammation and bronchial hyperreactivity with the allergens in the local environment dictating the specificity of the antibody response.16 Triggers of acute asthma can be non-specific (cold air, exercise, atmospheric pollutants), specific allergens (housemite, pollen, animal danders), modifiers of airway control (aspirin, β-blockers) or stress and emotion. No precipitant can be identified in over 30% of patients.
PATHOPHYSIOLOGY
The postmortem airway pathology of patients who die from acute asthma includes bronchial wall thickening from oedema and inflammatory cell infiltrate, hypertrophy and hyperplasia of bronchial smooth muscle and submucosal glands, deposition of collagen beneath the epithelial basement membrane and prominent intraluminal secretions. These secretions may narrow or occlude the small airways and postmortem studies frequently report extensive plugging and atelectasis.17,18 When interpreting the latter findings it is important to remember that during life in very severe asthma, the lungs are hyperinflated close to total lung capacity (TLC) by maximal inspiratory effort and to beyond TLC during mechanical ventilation. At this time radiological atelectasis is rare, suggesting that most airways are communicating at these high lung volumes, even if airways are very narrowed. Only at death does the prolonged apnoea allow lung deflation, widespread airway closure and alveolar gas absorption to give the postmortem appearance of extensive complete occlusion that was not present to the same degree during life.
In some deaths bronchial mucus is absent; in these cases airway obstruction may be mainly due to intense smooth bronchoconstriction.
This observation may account for two patterns of progression of asthma:
CLINICAL FEATURES AND ASSESSMENT OF SEVERITY
Triage and assessment of severity of the acute asthma attack are crucial. Underestimation or non-measurement of asthma severity is associated with increased mortality.26,27 Assessment has two key features: assessment of initial severity and ongoing assessment of response to treatment.
HISTORY
Any history of prior intubation and mechanical ventilation for asthma is a predictor for life-threatening asthma.26,27 A history of poor asthma control and multiple recent medical presentations for asthma are recognised risk factors. Other risk factors include a poor response to prior treatments and poor psychosocial circumstances.
PHYSICAL EXAMINATION
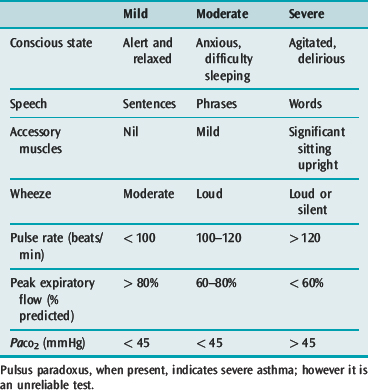
The general appearance and level of distress can be an important indicator of severity (Table 31.1). Use of accessory muscles, suprasternal retraction, markedly diminished breath sounds or a silent chest, central cyanosis, inability to speak (sentences, phrases, single words), a disturbance in the level of consciousness, upright posture and diaphoresis all suggest a severe attack.28 A respiratory rate > 30 breaths/min, pulse rate > 120 beats/min and pulsus paradoxus of > 15 mmHg are associated with severe asthma, though their absence does not preclude life-threatening asthma.
VENTILATORY FUNCTION TESTS
The patient may not be able to perform these due to breathlessness. However, forced expiratory volume in 1 second (FEV1) and peak expiratory flow rate (PEFR) are useful indicators of severity and response to treatment when done serially. An FEV1 < 1.0 litre or PEFR < 100 l/min indicates very severe asthma at significant risk of requiring mechanical ventilation. In some patients forced expiration may worsen symptoms; these measurements should cease if this occurs.29
PULSE OXIMETRY
Pulse oximetry (SpO2) is usually readily available and provides a rapid assessment of oxygenation. It is also very valuable in regulation oxygen therapy, both for avoiding hypoxia (SpO2 > 90%)30 and avoiding potential side-effects of hyperoxia (SpO2 < 95%).31 Of course, pulse oximetry cannot assess arterial PCO2 or acid–base state or lactate and hence does not replace the need for blood gases
ARTERIAL BLOOD GASES
Arterial hypoxaemia is almost invariably present in a patient with severe asthma breathing room air, though it usually responds well to low-level oxygen supplementation (28–35%).30 Blood gases should not delay initiation of treatment, and are not required in mild asthma or moderate asthma that is responding well to treatment. They are very important in severe asthma, or moderate asthma with inadequate treatment response. The arterial PaCO2 is an important measure of severity and, if hypercapnia is present, an important guide to treatment response.
Ventilation is initially increased in an acute attack, leading to hypocapnia and a respiratory alkalosis.30 As the asthmatic attack worsens, the work of breathing, V/Q mismatch and adverse cardiopulmonary interactions all increase, and the minute ventilation required to maintain the same alveolar ventilation and PaCO2 increases. Eventually the patient is incapable of meeting this demand and the PaCO2 rises. The presence of hypercapnic acidosis is associated with an FEV1 of < 20% predicted and reliably indicates that asthma is severe. A metabolic acidosis may also be present, and this is most commonly due to lactic acidosis32 associated with intravenous or continuous nebulised β-agonists.33,34
CHEST X-RAY
Although not generally helpful in assessing severity,35 a chest X-ray should be performed when asthma is severe or refractory to treatment, when barotrauma or lower respiratory tract infection is suspected or when the diagnosis is in doubt. It is not required in milder attacks that respond well to treatment.
ASSESSMENT OF TREATMENT RESPONSE
Repeated evaluation of the patient’s response to treatment is a valuable tool in assessing severity of acute asthma. The response to the first 2 hours of treatment is an important predictor of outcome.36 Adverse events are associated with underestimation of severity, inadequate observation after initial assessment and under treatment. Ongoing evaluation of response to therapy is critical. This evaluation should include repeated assessments of:
MANAGEMENT
ESTABLISHED TREATMENTS
Initial therapy of acute severe asthma should include the following.
OXYGEN
Hypoxaemia contributes to life-threatening events that complicate acute severe asthma.37 Humidified supplemental oxygen should be titrated to achieve a SpO2 > 90%. The risk of oxygen-induced increasing hypercapnia with coexisting chronic obstructive pulmonary disease or pre-existing chronic hypercapnia is a well-known reason to maintain a lower SpO2 (e.g. 90–92%). This phenomenon was not believed to be relevant to the majority of patients with acute asthma; however, there is now emerging evidence that hyperoxia may be harmful to a more widespread group38 by releasing pulmonary hypoxic vasoconstriction, worsening V/Q matching and increasing hypercapnia. A recent randomised controlled trial31 showed a decrease in PaCO2 in a group receiving 28% O2 and an increase in PaCO2 in a group receiving 100% O2.
β-AGONISTS
Short-acting β-agonists remain the first-line bronchodilator therapy of choice.6,39–41 Agents include salbutamol (albuterol), terbutaline, isoprenaline and epinephrine. Salbutamol is generally the agent of first choice as it has relative β2-selectivity, with decreased β1-mediated cardiac toxicity. Long-acting β-agonists such as salmeterol have no role in status asthmaticus due to slow onset of action and association with fatalities in this setting.14 Eformotarol is a combined long- and short-acting β-agonist which could theoretically be used for acute asthma; however it is in dry powder form and unlikely to be helpful in intubated and mechanically ventilated patients. β-agonists cause bronchodilatation by stimulation of β2-receptors on airway smooth muscle and may reduce bronchial mucosal oedema.42
β-agonists are best given by metered-dose inhaler (MDI) and a spacer device. There are data to suggest that, in non-intubated patients, MDIs combined with a spacer device are more effective than nebulisers and are cheaper to use.43,44 In intubated patients both nebulisers and MDIs have been used effectively.45 The dose per MDI inhalation is 100 μg and this can be given, in severe asthma, in repeated doses at 1–5-minute intervals.
Alternatively, β-agonists can be given by nebuliser in high and repeated doses.46 The typical adult dose of salbutamol is 5–10 mg (in 2.5–5.0 ml diluent volume) every 2–4 hours but more frequent doses with a higher total dose are often required in severe asthma. It should be noted that less than 10% of the nebulised drug reaches the lung even under ideal conditions.47 Continuous nebulisation appears to be superior to intermittent doses and is commonly used at the beginning of treatment in severe asthma.14,48 The nebuliser should be driven by oxygen with the flow at 10–12 l/min and a reservoir volume of 2–4 ml so as to produce particles in the desired 1–3 μm range.49 The total dose should be modulated by response to treatment and the level of toxic side-effects.
Two-thirds of patients presenting acutely will respond well to inhaled β-agonists,50 irrespective of the method of administration. The remaining one-third are refractory even to high doses and usually require longer periods of intense treatment, including multiple other agents.
Intravenous β-agonists remain controversial. There is no clear evidence of benefit51 and significant side-effects. Despite this, intravenous β-agonists have a theoretical benefit of additional access to lung units with severe airflow obstruction and poor nebulised drug delivery, and some studies have demonstrated improved response when intravenous β-agonist is used.52 Intravenous β-agonists continue to be considered if the patient is not responding to continuous nebulisation.53 The typical dose is 5–20 μg/min but doses > 10 μg/min should be used with caution because of side-effects, which should be monitored closely. Salbutamol 100–300 μg may also be given intravenously to non-intubated patients in extremis or delivered down an endotracheal tube if there is not time to gain intravenous access.
Side-effects of β-agonists include tachycardia, arrhythmias, hypertension, hypotension, tremor, hypokalaemia, worsening of ventilation–perfusion mismatch and hyperglycaemia,54 but the most common side-effect of parenteral β-agonist – also occasionally seen with continuous nebulised β-agonists – is lactic acidosis. This occurs in over 70% of patients, has an onset within 2–4 hours of commencing an infusion or following an intravenous statim dose, levels may reach 4–12 mmol/l and may significantly add to respiratory acidosis and respiratory distress.34,55,56 Parenteral infusions should be initially limited to 10 μg/min and statim doses should not exceed 250 μg. Serum bicarbonate and lactate should be regularly monitored. If lactic acidosis becomes significant, the salbutamol infusion should be reduced or ceased. Lactic acidosis will generally resolve within 4–6 hours of infusion cessation and is seldom a problem with infusions in place for more than 24 hours.
Long-term high-dose β-agonist use has been associated with increased mortality,57 but whether high-dose β-agonists are a marker of disease severity, an indicator of suboptimal inhaled steroids or a direct cause of death is unclear. These concerns do not apply in the treatment of the acute asthma attack.
ANTICHOLINERGICS
Anticholinergics cause bronchodilatation by decreasing parasympathetic-mediated cholinergic bronchomotor tone.58 Ipratropium bromide is the most commonly used anticholinergic for asthma and is a quaternary derivative of atropine. A number of studies and meta-analyses now suggest clear additional benefit and few side-effects when ipratropium bromide is added to the β-agonist regimen59,60 and ipratropium is now considered accepted first-line therapy for acute severe asthma in conjunction with β-agonist therapy. Preservative-induced bronchoconstriction has been reported in a few patients and can be prevented by using preservative-free solutions.61 The bronchodilatation effect of ipratropium bromide appeared to be maximal with a dose of 250 μg when studied in children between 9 and 17 years of age. The optimal dose is not known in adults; a reasonable regimen would be to add 500 μg ipratropium bromide to the salbutamol nebuliser every 2–6 hours; however, initial dose intervals as low as 10–20 minutes have been recommended.62
CORTICOSTEROIDS
The role of corticosteroids in the acute asthma attack has been well established. Systemic steroids should be considered in all but mild exacerbations of asthma.41 Their benefits include increased β-responsiveness of airway smooth muscle, decreased inflammatory cell response and decreased mucus secretion. Early treatment with corticosteroids has been shown to decrease the likelihood of hospitalisation and decrease the mortality rate from acute asthma. Systematic reviews41,63 suggest that effects commence within 6–12 hours, that oral administration is as effective as intravenous and that there is little evidence of benefit for initial daily doses exceeding 800 mg/day hydrocortisone (160 mg/day methylprednisolone) given in four divided doses.
Inhaled steroids have established long-term benefit and are believed to be a major factor in asthma mortality reduction.1–3 There is now emerging evidence that inhaled steroids may also have a role during an acute attack64,65 and it appears reasonable to use them routinely from day 1 as they may also enable more rapid dose reduction of parenteral steroids, potentially reducing side-effects.
Side-effects of corticosteroids include hyperglycaemia, hypokalaemia, hypertension, acute psychosis and myopathy,66,67 though they are usually well tolerated acutely. The immunosuppressive effects can increase the risk of infections, including Legionella, Pneumocystis carinii and varicella,68,69 especially when the patient is on long-term corticosteroids. Allergic reactions including anaphylaxis have been reported with the use of most corticosteroid preparations.
AMINOPHYLLINE
There have been conflicting reports regarding the efficacy of aminophylline in acute asthma ranging from no benefit70 to improved lung function and improved outcome.71 However it is accepted that aminophylline is an inferior bronchodilator, with a narrow therapeutic range and frequent side-effects,72 including headache, nausea, vomiting and restlessness; cardiac arrhythmias and convulsions can occur at serum levels above 200 μmol/l (40 mg/l).
As a result, aminophylline is not a first-line treatment.40,41,53 Aminophylline may be given to patients with acute asthma who are not showing a favourable response to full treatment with first-line agents. Careful administration and monitoring are required with an initial loading dose of 3 mg/kg (maximum 6 mg/kg, omitted if the patient is already taking oral theophylline) and an infusion of 0.5 mg/kg per hour. This should be reduced in patients with cirrhosis, cardiac failure or chronic obstructive pulmonary disease and in patients taking cimetidine, erythromycin or antiviral vaccines. Drug levels should be taken after a loading dose (if given), and then 24 hours later, aiming for a level of 30–80 μmol/l (5–12 mg/l). Levels should be repeated daily thereafter until stability has been achieved. The duration should be based on the response to treatment.
NON-ESTABLISHED TREATMENTS
EPINEPHRINE
Epinephrine has some theoretical advantages over pure β2-agonists in that its additional α-agonist actions of vasoconstriction and mucosal shrinkage may improve airway calibre. However, in practice, nebulised or subcutaneous epinephrine has not been shown to confer any advantage over nebulised β2-agonists and is not recommended because of its cardiac side-effects.62 Epinephrine may be tried in the patient who is failing to respond to conventional treatment. The nebulised dose is 2–4 mg in 2–4 ml (1% solution, 0.05 ml/kg) 1–4-hourly. The subcutaneous dose is 0.2–0.5 mg (0.2–0.5 ml of 1:1000 epinephrine) repeated if necessary 2–3 times at 30-minute intervals. Epinephrine by infusion may avert mechanical ventilation in very severe cases but should be used with caution with electrocardiogram monitoring and preferably with central venous access. An initial intravenous dose of 0.2–1.0 mg (2–10 ml of 1:10 000 epinephrine) is given slowly over 3–5 minutes. This may be followed by a continuous infusion of 1–20 μg/min, which is weaned when the acute attack subsides.73


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


