Chapter 15 Lactic acidosis
Lactic acidosis is defined by convention as the combination of an increased blood lactate concentration (> 5 mmol/l) and acidaemia (arterial blood pH of < 7.35).1,2 However hyperlactaemia occurs whenever the blood lactate concentration is above the normal range (< 2 mmol/l). Lactic acidosis may be masked by coincident metabolic or respiratory alkalosis. Critically ill patients with lactic acidosis usually have a high mortality3 and blood lactate concentrations > 8 mmol/l predict fatality.4 A prospective study reported 83% mortality in patients with blood lactate concentrations of 10 mmol/l.1 In each individual patient, however, prognosis is completely dependent on the underlying condition, with the initial degree of lactic acidosis being a clinically useful indicator of shock severity, and serial assessment allowing evaluation of response to therapy. Recently, blood lactate concentration has been identified as one marker of the degree of shock triggering treatment in septic patients who may benefit from early goal-directed therapy.5 It is important to remember that, in healthy athletes, severe lactic acidosis during exercise is a normal, self-limited observation.
PATHOPHYSIOLOGY
There is a continuous cycle of lactate production and metabolism. Lactate is produced at about 0.8 mmol/kg per h, while simultaneous metabolism in liver, kidneys, skeletal muscle, brain and red blood cells ensures that blood lactate concentrations are normally low (< 1 mmol/l). Lactic acidosis occurs when lactate production exceeds metabolic capacity, or when metabolic capacity is decreased by organ dysfunction. The liver has a key role in lactate homeostasis and many patients who develop lactic acidosis have decreased metabolic capacity due to liver disease.Glucose is derived from absorption, glycogen and gluconeogenesis. Rate of glycolysis is controlled by three unidirectional enzymes and the activity of 6
Formation and metabolism of lactate in cells are catalysed by lactate dehydrogenase (LDH):
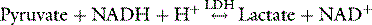
Lactate formation is in part dependent upon pyruvate concentrations and pyruvate is sourced from glycolysis (85%) and proteolysis (15%). one of these enzymes is increased by increasing intracellular pH. Acidosis therefore decreases (and alkalosis increases) glycolysis and pyruvate, and consequently lactate production. In oxygen excess, pyruvate is oxidised and lactate does not accumulate. Anaerobic metabolism however, causes lactate accumulation and an increased lactate/pyruvate ratio. The blood lactate/pyruvate ratio is however a poor indicator of mitochondrial concentrations, and is not of clinical use.
CLASSIFICATION (TYPES A AND B)
The Cohen and Woods classification of lactic acidosis7 defines two subgroups depending on the presence (type A) or absence (type B) of tissue hypoxia (Table 15.1). Type A lactic acidosis due to tissue hypoxia is common in critically ill patients. Type B (no tissue hypoxia) is much less common. Some patients with type B lactic acidosis have increased lactate generation (some malignancies and toxins impair oxygen utilisation by cells, despite adequate oxygen delivery), and other patients have decreased lactate clearance (liver disease). Patients with liver disease have greater hyperlactaemia during states of increased lactate production than those with normal liver function.6
Table 15.1 Classification of lactic acidosis
| Type A | Shock |
| Very severe hypoxaemia | |
| Very severe anaemia | |
| Carbon monoxide poisoning | |
| Type B1 (underlying disease) | Sepsis |
| Liver failure | |
| Thiamine deficiency | |
| Malignancy | |
| Phaeochromocytoma | |
| Diabetes | |
| Type B2 (drug or toxin) | Epinephrine |
| Salbutamol | |
| Propofol | |
| Nucleoside analogue reverse transcriptase inhibitor | |
| Ethanol | |
| Methanol | |
| Paracetamol | |
| Nitroprusside | |
| Salicylates | |
| Ethylene (and propylene) glycol | |
| Biguanides | |
| Fructose | |
| Sorbitol | |
| Xylitol | |
| Cyanide | |
| Isoniazid | |
| Type B3 (rare inborn errors of metabolism) | Glucose-6-phosphatase deficiency |
| Fructose-1,6 diphosphatase deficiency | |
| Pyruvate carboxylase deficiency | |
| Deficiency of enzymes of oxidative phosphorylation |
COMBINED ABNORMALITIES (TYPES A AND B)
In clinical practice, separation of types A and B lactic acidosis is usually not helpful, because many critically ill patients have combined abnormalities. Increased lactate formation from tissue hypoxia and decreased lactate clearance often occur together. In patients with cancer, anaerobic glycolysis may be increased while hepatic lactate metabolism is impaired by tumour replacement. Diabetic patients may present with shock, but in non-insulin-dependent diabetes there may also be a defect in pyruvate oxidation, and in diabetic ketoacidosis ketones may also inhibit hepatic lactate uptake. Thiamine and biotin are essential cofactors for pyruvate dehydrogenase activity and for conversion of pyruvate to oxaloacetate. Malnutrition (beri-beri) and inadequate parenteral nutrition have therefore been associated with lactic acidosis due to deficiencies of these cofactors. In these cases, pyruvate accumulation increases lactate production. In alcoholics, ethanol oxidation increases the conversion of pyruvate to lactate and inhibits other pathways of pyruvate metabolism. Phenformin therapy is associated with lactic acidosis for several reasons: phenformin increases glycolysis in peripheral tissues, inhibits pyruvate oxidation, increases splanchnic lactate production and decreases hepatic lactate clearance. Phenformin was therefore used to induce lactic acidosis in older animal models until it was recognised that phenformin is also a potent cardiac depressant. These studies reporting the effects of lactic acidosis on cardiac function were flawed. Both endogenous and infused catecholamines cause hepatic vasoconstriction and impair hepatic lactate clearance, and epinephrine also increases hepatic glycogenolysis to lactate.8 Importantly, acidosis also stimulates adrenal catecholamine release, which may mask other cardiac effects of acidosis.
SEPSIS
There has been much recent debate9 whether the hyperlactaemia of sepsis results from net increased cellular production10 or reduced net clearance.11 However the potential for sepsis to derange lactate metabolism is clear. Both impaired regional microvascular and mitochondrial dysfunction have been implicated in the pathogenesis of lactic acidosis during sepsis. Excess catecholamines may impair hepatic lactate extraction (by reducing regional hepatic blood flow) and increase lactate production (increased glycogenolysis). At the same time lactate clearance is decreased because pyruvate dehydrogenase activity is reduced in both skeletal muscle and liver. Mitochondrial pyruvate oxidation is impaired.
Tissue hypoxia may not be a major mechanism for regional lactate production during sepsis: hyperlactaemia is thought to be linked to the severity of the septic cellular inflammatory response and hypermetabolic state.9,12 Net lactate production from the hepatosplanchnic bed is uncommon in septic patients13 and nuclear magnetic resonance spectroscopy suggests that hyperlactaemia may occur without tissue hypoxia.14
LUNG INJURY
The lung is a primary source of lactate production in patients with acute lung injury, with pulmonary release of lactate being directly related to the severity of lung injury,15,16 supporting the view that the primary contributors are the tissues with the most inflammation or injury. The increased lactate production by the injured lung is not only secondary to anaerobic metabolism in the hypoxic regions of the lung but also may be due to altered glucose metabolism and a direct effect of cytokines on pulmonary cells.16
Recent laboratory data suggest that both metabolic and respiratory acidosis protect the lung against injury whereas correction of acidosis compounded the injury.17,18 The two ventilator trials19,20 which demonstrated a positive impact on mortality in acute respiratory distress syndrome (ARDS) by limiting tidal volume and airway pressures differed widely on how they regarded the resultant hypercapnic acidosis. While Amato et al.19 allowed elevation of the PaCO2 (permissive hypercapnia) and resultant acidosis, the ARDSnet group20 in contrast aggressively corrected the hypercapnic acidosis by increasing the respiratory rate and allowing administration of sodium bicarbonate. There is growing evidence that not only may hypercapnic acidosis be beneficial in lung injury,17,21 but the ARDSnet interventions aimed at correcting the acidosis may also be deleterious.18,22 These findings have not only promoted a greater tolerance of acidosis in ARDS but also increased reluctance to buffer the acidosis exogenously towards ‘normal’ values.
ASTHMA
Lactic acidosis also often occurs in patients with acute severe asthma.23 Fatiguing respiratory muscles have been implicated, but severe lactic acidosis also occurs in sedated, paralysed mechanically ventilated patients who have no endogenous respiratory muscle activity.24 β-agonists, including salbutamol and epinephrine cause lactic acidosis by increasing gluconeogenesis, glycogenolysis, lipolysis and cyclic adenosine monophospate (AMP) activity. Clinical experience suggests that infusions of β-agonists are the primary cause of lactic acidosis in patients with asthma because decreasing intravenous salbutamol infusions to less than 10 mcg/min is usually associated with resolution of the acidosis. In asthma, lactic acidosis does not have specific prognostic implications.
CARDIAC SURGICAL PATIENTS
Hyperlactaemia during cardiopulmonary bypass is relatively frequent and is associated with an increased postoperative morbidity.25 Recent work has suggested that this ‘on-pump’ hyperlactaemia is secondary to inadequate peripheral oxygen delivery (DO2) which creates a condition similar to cardiogenic shock, leading to both direct lactate formation by dysoxic tissues and also to catecholamine release, insulin resistance and hyperglycemia-induced lactate production.
The use of epinephrine after cardiopulmonary bypass precipitates lactic acidosis in some patients.26 This phenomenon is probably β-agonist-mediated, is associated with increased whole-body blood flow and resolves after substitution of norepinephrine. However there is emerging evidence that the severity of lactic acidosis following cardiac surgery is related to certain genetic polymorphisms in tumour necrosis factor and interleukin-10 genes.27 Similar to asthma, lactic acidosis associated with the administration of epinephrine in this setting does not have the adverse implications of lactic acidosis associated with shock.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


