Chapter 81
Acute Kidney Injury and Rhabdomyolysis
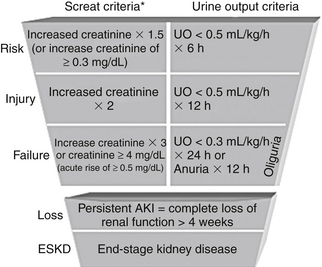
Acute kidney injury (AKI) is an abrupt decline in renal function manifested by increased plasma creatinine and increased blood urea nitrogen (BUN) concentrations and a declining urine output. In 2004, the Acute Dialysis Quality Initiative defined AKI using the acronym RIFLE (Figure 81.1) to represent worsening renal function classes. Each letter in RIFLE represents, respectively, risk, injury, failure, and loss of renal function, and ultimately “E” stands for end-stage renal disease (ESRD). The RIFLE criteria deem a small increase in the serum creatinine concentration of more than 0.3 mg/dL over one to several days as clinically significant. The frequency of AKI ranges from 2% to 5% in hospitalized general medical-surgical patients to 10% to 25% in patients in the intensive care unit (ICU). The RIFLE criteria are a sensitive definition of acute renal failure, allowing physicians to recognize the increased morbidity and mortality associated with even slight serum creatinine elevations. Although AKI frequently develops as a direct complication of the patient’s underlying disease process, the majority of episodes are related to medical care.
Differential Diagnosis
Prerenal Acute Kidney Injury (AKI)
Any decrease in renal perfusion activates physiologic processes designed to preserve glomerular filtration rate (GFR) and solute excretion. Moderate decreases in perfusion stimulate both neural and hormonal factors (primarily angiotensin II, prostaglandins, catecholamines, aldosterone, and vasopressin), producing selective postglomerular (efferent) arteriolar vasoconstriction. This efferent vasoconstriction maintains GFR and enhances renal sodium reabsorption. However, with more severe and prolonged renal hypoperfusion, these regulatory processes fail to maintain normal GFR, culminating in prerenal AKI (Table 81.1A) and nitrogenous waste accumulation in the blood.
TABLE 81.1A
Causes of Prerenal Acute Kidney Injury
| Volume Depletion | Renal Vasoconstriction |
| Decreased effective arterial blood volume | Cyclosporine |
| Congestive heart failure | Hepatorenal syndrome |
| Cirrhosis | Hypercalcemia |
| Nephrotic syndrome | Nonsteroidal anti-inflammatory drugs (NSAIDs) |
| Tacrolimus |
True intravascular volume depletion or states depleting effective arteriolar volume (for example, congestive heart failure, cirrhosis, or nephrotic syndrome) can lead to prerenal AKI. In these latter states, diuretic therapy may further compromise renal perfusion by superimposing true volume depletion. Drugs that block homeostatic responses and the aforementioned autoregulatory mechanisms to renal hypoperfusion (angiotensin-converting enzyme inhibitors [ACEIs], angiotensin-receptor blockers [ARBs], and nonsteroidal anti-inflammatory drugs [NSAIDs]) may worsen renal function. Vasoconstrictors (such as catecholamines, cyclosporine, or tacrolimus) potentially decrease renal perfusion and exacerbate prerenal AKI. The hepatorenal syndrome (defined in Table 81.2) results from severe renal vasoconstriction associated with end-stage liver disease (see Chapter 27) and does not improve with volume loading. In the critically ill patient, multiple factors usually contribute to prerenal AKI.

TABLE 81.2
Diagnostic Hepatorenal Syndrome Criteria in Cirrhosis
Cirrhosis with ascites
Serum creatinine > 1.5 mg/dL
No improvement of serum creatinine (decrease to a level of < 1.5 after at least 2 days with diuretic withdrawal and volume expansion with albumin; the recommended dose of albumin is 1 g/kg of body weight per day up to maximum of 100 g/day
Absence of shock
No current or recent treatment with nephrotoxic drugs
Absence of parenchymal kidney disease as indicated by proteinuria > 500 mg/day, microhematuria > 50 RBCs per hpf or abnormal renal ultrasound
RBCs, red blood cells; hpf, high-powered field.
From Salerno F, Gerbes A, Gines P, et al: Diagnosis, prevention and treatment of hepatorenal syndrome in cirrhosis. Postgrad Med J 84:662-670, 2008.
The hallmarks of prerenal AKI are the excretion of a concentrated urine (urine osmolality > 700 mOsm/kg, urine specific gravity > 1.020), with relatively low sodium concentrations (urine sodium < 20 mEq/L [< 20 mmol/L]), fractional excretion of sodium (FENA) less than 1% (see Chapter 39), and rapid reversibility with correction of the underlying cause. FENA should be used cautiously as a strict diagnostic criterion (Table 81.1B), as patients with CKD may be unable to concentrate their urine or achieve a FENA less than 1%. Furthermore, it is extremely important to only use FENA in the setting of oliguria (e.g., < 500 mL urine output in 24 hours). Diuretics can also produce a FENA > 1% in patients with prerenal AKI.
Conversely, conditions not defined as prerenal AKI (such as contrast nephropathy, rhabdomyolysis, hemoglobinuria, and urinary tract obstruction) may also have a low FENA (see Table 81.1B).
Postrenal Acute Kidney Injury (AKI)
Obstruction to urine flow at any level of the urinary collecting system may produce AKI (Table 81.3). Lower urinary tract obstruction may occur at the level of the bladder, bladder outlet, or urethra. Upper urinary tract obstruction may occur at the level of the ureter or the renal pelvis. Upper tract obstruction must be bilateral to cause AKI, absent a single functioning kidney or baseline renal insufficiency. Complete obstruction produces anuria, whereas partial obstruction yields variable urine output, with polyuria or fluctuation between polyuria and anuria characteristic.

Exclude postrenal causes in all patients with AKI, as obstructive renal failure is potentially reversible if promptly diagnosed and decompressed. Demonstrating residual bladder volume after voiding (postvoiding residual) > 100 mL by bladder catheterization is diagnostic of bladder outflow obstruction or a neurogenic bladder. Perform renal ultrasonography to rule out upper tract obstruction evidenced by hydronephrosis.
Intrinsic Acute Kidney Injury (AKI)
Intrinsic renal parenchymal injury producing AKI may be characterized into five broad categories on the basis of the underlying pathogenesis: (1) acute tubular necrosis (ATN), (2) acute interstitial nephritis (AIN), (3) acute glomerulonephritis (AGN), (4) intratubular obstruction, and (5) acute vascular syndromes (Table 81.4). Of these categories, ATN is by far the most common cause of intrinsic AKI.


Acute Tubular Necrosis (ATN)
Agents that can produce nephrotoxic ATN include aminoglycoside antibiotics, amphotericin B, acetaminophen, cisplatin, radiocontrast media, possibly free hemoglobin, and myoglobin. Contrast-induced nephropathy is the most common to cause ATN. In most ICU patients, ATN is multifactorial, resulting from a combination of nephrotoxic and ischemic insults.
< div class='tao-gold-member'>

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


