Chapter 52
Acute Heart Failure Syndromes 
Since the 1980s, the incidence and prevalence of chronic heart failure have been increasing rapidly with estimates showing ∼1% to 2% of the U.S. population being affected, predominantly in patients older than 65 years. This increase can be attributed to a combination of an aging population with increasing incidences of risk factors for heart failure (e.g., hypertension and diabetes), improved survival postmyocardial infarction (Chapter 50), and improved prevention of sudden cardiac death. The result is a higher prevalence of patients living with, rather than dying from, myocardial dysfunction.
Prognosis
Prognosis of AHFS can be divided into three distinct categories: in-hospital (inpatient) mortality, postdischarge morbidity and mortality, and longer-term morbidity and mortality. Inpatient mortality rates for patients with AHFS, excluding the cardiogenic shock subset (discussed in Chapter 8), typically range from 3% to 7%. Large registry data sets from the United States have identified several key factors as predictors of in-hospital mortality. The Acute Decompensated Heart Failure National Registry (ADHERE identified three variables present on admission as associated with in-hospital mortality: blood urea nitrogen (BUN) > 43 mg/dL, lower systolic blood pressure (< 115 mm Hg), and elevated serum creatinine (> 2.75 mg/dL). A separate registry, the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) registry, also identified serum creatinine and lower systolic blood pressure (SBP) as predictors of in-hospital mortality with the addition of several other variables (age, heart rate, presence of LV dysfunction and serum sodium). Subsequent analyses have also shown several other variables to be associated with in-hospital mortality including elevated brain natriuretic peptide (BNP), elevated troponin, and the need for inotropic therapy. Despite adequate symptom relief during the inpatient hospitalization, postdischarge readmission and mortality rates remain high (10% to 20% and 20% to 30%, respectively). Multiple variables associated with short-term readmissions and mortality have been identified, including systolic blood pressure, admission creatinine and hemoglobin, tachycardia, prolonged QRS, new onset arrhythmia during admission, worsening renal function during or after hospitalization, discharge hyponatremia, and discharge use of evidence-based therapies (angiotensin-converting enzyme [ACE]-I inhibitors, angiotensin receptor blockers [ARBs], beta-blockers) among others. Regarding long-term mortality, the data are not as robust but show that each hospitalization for acute heart failure (AHF) negatively impacts prognosis in an additive fashion ( Figure 52.E1). Not surprisingly, variables identified as having an impact on long-term prognosis in chronic heart failure, such as elevated BNP, QRS duration, and kidney dysfunction, have been shown to affect long-term prognosis after an episode of AHF.
Figure 52.E1). Not surprisingly, variables identified as having an impact on long-term prognosis in chronic heart failure, such as elevated BNP, QRS duration, and kidney dysfunction, have been shown to affect long-term prognosis after an episode of AHF.
Pathophysiology
Cardiac Output and Mean Arterial Pressure
Cardiac output (cardiac index is normalized for body surface area [BSA]) expressed in liters of blood per minute is determined by the product of stroke volume and heart rate (see Equation 1 in this chapter and Table 8.1 in Chapter 8). The heart rate is determined by the patient’s underlying electrical rhythm and balance of sympathetic and parasympathetic inputs, which in turn are often driven by the patient’s clinical status. The stroke volume is determined by the three principal factors: preload, intrinsic myocardial contractility, and afterload (see Figures 8.2 and 8.3 in Chapter 8). 
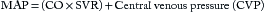
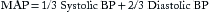
or
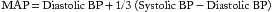
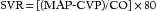
In systolic heart failure, an inciting event (e.g., myocardial infarction, myocarditis, hypertensive crisis) results in myocardial dysfunction. This dysfunction results in activation of numerous compensatory mechanisms in an attempt to maintain cardiac output and tissue perfusion. One of the first mechanisms invoked is an increase in sympathetic activation with a concomitant decrease in parasympathetic activity. This results in higher norepinephrine levels that, in the short term, increase CO via increasing contractility and heart rate and maintain blood pressure via both increasing CO as well as inducing peripheral vasoconstriction. The increased sympathetic activation in addition to other stimuli also stimulates activation of the renin-angiotensin system (RAS). Stimulation of the RAS induces sodium reabsorption in the kidney, peripheral vasoconstriction, and aldosterone secretion as well as positive feedback to further increase sympathetic activation, all of which function to maintain CO in the short term. This leads to salt and water retention and increased preload. Counter regulatory mechanisms, such as secretion of brain and atrial natriuretic peptides (BNP and ANP), are invoked in an attempt to counteract the effects of RAS activation. However, these mechanisms appear to be less effective in patients with heart failure.
In Figure 8.2, the classic “Frank-Starling curves” demonstrate that with increasing left ventricular filling pressure (LVEDP), or preload, there is a concomitant increase in cardiac output via the mechanism of increasing stroke volume. This relationship is maintained at low, normal, and mildly elevated filling pressures with subsequent flattening of the slope of the curves at higher filling pressures indicating a “limit” to the ability of the heart to augment stroke volume at the extremes of filling pressures. Whereas line A represents normal myocardial contractility, line C represents depressed myocardial functioning with subsequent lower cardiac outputs at any given filling pressure as well as a flatter slope indicating a decreased ability to augment stroke volume in the setting of myocardial dysfunction. It is important to note that although the slope of the line flattens, there is no descending limb to the curves indicating decreasing stroke volumes at excessively high filling pressures. However, these studies were performed in isolated myocytes rather than in whole hearts. Although myocytes continue to increase their contractility with increasing “stretch” or filling pressures, elevated filling pressures exert a negative impact on global left ventricular (LV) functioning. At high filling pressures, there is neurohormonal activation, distortion of LV architecture, stretching of the papillary muscles, and subsequent induction of functional mitral regurgitation that leads to the phenomenon colloquially termed “falling off the starling curve.” Figure 8.3 illustrates a similar but opposite relationship between afterload and SV showing that with increasing afterload, SV decreases. Importantly, curve C, which again represents depressed myocardial contractility, shows a significantly steeper slope indicating a more pronounced negative effect of afterload on SV and CO in the setting of myocardial dysfunction.
Pathophysiology of Acute Heart Failure
Multiple precipitating factors have been identified for AHFS (Box 52.E1).  All of these factors lead to either increased volume and congestion or decreased myocardial systolic or diastolic performance. Once the inciting event occurs, neurohormonal activation is increased, leading to increased myocardial oxygen demand (and possible ischemia resulting from supply/demand mismatch [Chapter 50], peripheral vasoconstriction, and sodium and water retention in a positive feedback loop. If left uncorrected, these changes lead to further decrements in myocardial function, organ hypoperfusion, and eventual cardiogenic shock (Chapter 8). Although this cycle pertains to all patients with AHFS, patients with obstructive coronary artery disease are particularly susceptible to the development of ischemia secondary to supply-demand mismatch in the setting of increased myocardial oxygen demand with concomitant drops in coronary perfusion pressures (as a result of either hypotension or elevated LV diastolic pressures).
All of these factors lead to either increased volume and congestion or decreased myocardial systolic or diastolic performance. Once the inciting event occurs, neurohormonal activation is increased, leading to increased myocardial oxygen demand (and possible ischemia resulting from supply/demand mismatch [Chapter 50], peripheral vasoconstriction, and sodium and water retention in a positive feedback loop. If left uncorrected, these changes lead to further decrements in myocardial function, organ hypoperfusion, and eventual cardiogenic shock (Chapter 8). Although this cycle pertains to all patients with AHFS, patients with obstructive coronary artery disease are particularly susceptible to the development of ischemia secondary to supply-demand mismatch in the setting of increased myocardial oxygen demand with concomitant drops in coronary perfusion pressures (as a result of either hypotension or elevated LV diastolic pressures).
Clinical Presentation and Initial Assessment
Evaluation of any patient with known or suspected cardiac dysfunction involves a careful history and physical examination focusing on three vital components: (1) assessing volume status, (2) assessing tissue perfusion, and (3) evaluating for precipitating factors and comorbidities.
A typical approach to the initial assessment is depicted in Figure 52.1 in which patients are grouped into four hemodynamic profiles. The patients in profile A are euvolemic and well perfused and represent well-compensated outpatients, management of whom is outside the scope of this chapter. Based on registry data in the United States and Europe, the majority of patients admitted for AHF present with symptoms of congestion  (Table 52.E1), in particular dyspnea as represented by profiles B and C. As patients with chronic heart failure develop multiple adaptive responses to chronically elevated filling pressures, congestion is often under-recognized on examination. The sensitivities of findings commonly associated with congestion such as rales, edema, or an abnormal chest chest radiograph have sensitivities of < 30% for detecting congestion in patients with chronic heart failure. In contrast, an elevated jugular venous pressure (JVP) or positive hepatojugular reflux, which in the absence of pulmonary hypertension or right-sided heart failure mirrors left-sided filling pressures, has a sensitivity of 80% for detecting a pulmonary capillary wedge pressure of > 18 mm Hg.
(Table 52.E1), in particular dyspnea as represented by profiles B and C. As patients with chronic heart failure develop multiple adaptive responses to chronically elevated filling pressures, congestion is often under-recognized on examination. The sensitivities of findings commonly associated with congestion such as rales, edema, or an abnormal chest chest radiograph have sensitivities of < 30% for detecting congestion in patients with chronic heart failure. In contrast, an elevated jugular venous pressure (JVP) or positive hepatojugular reflux, which in the absence of pulmonary hypertension or right-sided heart failure mirrors left-sided filling pressures, has a sensitivity of 80% for detecting a pulmonary capillary wedge pressure of > 18 mm Hg.
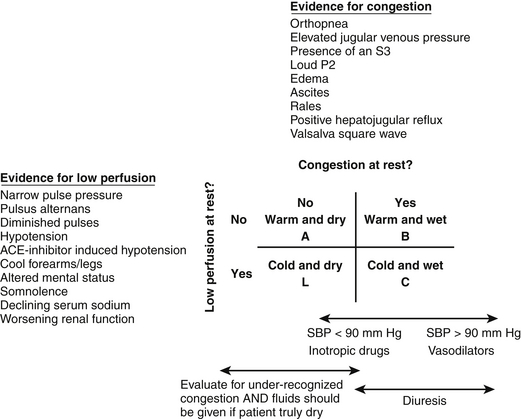
Profiles C and L in Figure 52.1 represent the patients with low perfusion with and without congestion, respectively. Low output heart failure and in particular cardiogenic shock patients constitute a minority of patients admitted with AHF. In contrast to patients with congestion who present with the typical symptoms listed in  Table 52.E1, patients with hypoperfusion or low output heart failure typically present with more nonspecific symptoms such as fatigue and lethargy. The constellation of vague symptoms, especially if combined with symptoms less classically thought to be due to heart failure such as abdominal pain and bloating, often lead to the misdiagnosis or delayed diagnosis of the low output state. Other historical or laboratory clues to low perfusion include new diuretic resistance as an outpatient, hyponatremia, elevated creatinine, or abnormal liver function tests as described later. In addition, blood pressure and heart rate can be important clues to hypoperfusion. Sinus tachycardia in the absence of an alternate cause (pain, pulmonary edema, hypovolemia, etc.) is an ominous finding often indicating significantly depressed cardiac function with an inability to augment stroke volume and subsequent reliance on heart rate to maintain cardiac output (see Figure 52.E1).
Table 52.E1, patients with hypoperfusion or low output heart failure typically present with more nonspecific symptoms such as fatigue and lethargy. The constellation of vague symptoms, especially if combined with symptoms less classically thought to be due to heart failure such as abdominal pain and bloating, often lead to the misdiagnosis or delayed diagnosis of the low output state. Other historical or laboratory clues to low perfusion include new diuretic resistance as an outpatient, hyponatremia, elevated creatinine, or abnormal liver function tests as described later. In addition, blood pressure and heart rate can be important clues to hypoperfusion. Sinus tachycardia in the absence of an alternate cause (pain, pulmonary edema, hypovolemia, etc.) is an ominous finding often indicating significantly depressed cardiac function with an inability to augment stroke volume and subsequent reliance on heart rate to maintain cardiac output (see Figure 52.E1).
TABLE 52.E1
Signs and Symptoms of Congestion and Low Perfusion (also see Figure 52.1)
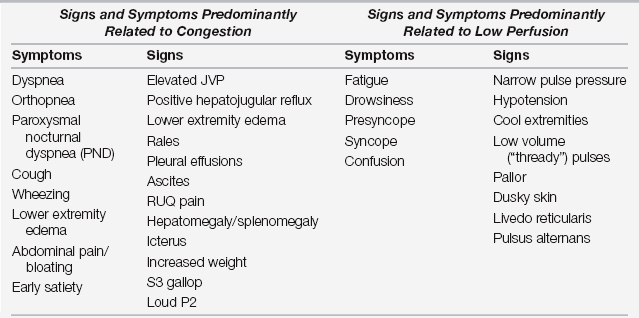
Adapted with modification from Braunwald E, Bonow R, Mann D, et al: Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. Philadelphia: Elsevier Saunders, 2008.
As part of the initial assessment, one should always attempt to identify a possible precipitant cause. Box 52.E1 lists the common precipitants of AHFS, the most common of which is medication or dietary nonadherence. In addition, use of medications such as nonsteroidal anti-inflammatory drugs (NSAIDs), certain calcium channel blockers, and other medications that can lead to volume retention are frequent causes of heart failure decompensation. Several of the precipitants, such as ischemia, arrhythmias, and pericardial tamponade (Chapter 54), need urgent or emergent treatment without which the AHFS will only progress in severity. Identifying a precipitating factor is not always possible but, if present, is imperative not only for directing in-hospital treatment but also for delivering appropriate patient counseling or medication adjustments on discharge.
Laboratory and Non-Invasive Testing
In addition to creatinine and BUN, other relevant laboratory values include serum sodium and potassium. Hyponatremia, also often a reflection of neurohormonal activation, is present in 25% of patients admitted with AHFS and portends a worse prognosis. Multiple medications commonly prescribed for patients with chronic heart failure such as angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), aldosterone antagonists, and diuretics can affect potassium levels, and hypo- or hyperkalemia should be promptly corrected. Mild elevations in liver enzymes are often seen as a result of right-sided congestion. However, markedly elevated enzymes indicate either hypoperfusion or primary liver injury including drug toxicity. A routine complete blood count should also be performed to rule out anemia and to aid in evaluating for the possibility of infection as a precipitating mechanism.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


