Chapter 16 Acute cardiac syndromes, investigations and interventions
Cardiovascular disease (CVD) accounts for 35–40%1 of deaths in western industrialised society, with coronary artery disease (CAD) being responsible for about half of these. In patients over age 40, acute myocardial infarction (MI) is the cause of approximately 20% of all deaths. Up to 80–90% of these deaths occur outside hospital. Of patients admitted to hospital, mortality is 8–9%,1 much higher in at-risk groups. Lowering in-hospital mortality from CAD requires us to identify rapidly patients who are at risk and to implement evidence-based treatment regimens.
MYOCARDIAL INFARCTION
Acute MI can occur in association with or result from a number of different pathological and epidemiological mechanisms:2
Each mechanism may have a different long-term prognosis despite similar biomarker and ECG changes. Such classifications may be clinically important when interpreting the results of clinical trials.2
ACUTE CORONARY SYNDROMES (ACS)
ACS represent the largest group of patients developing MI. They describe the spectrum of patients who present with chest discomfort or other symptoms caused by acute myocardial ischaemia (Figure 16.1). ACS can be further divided into acute MI and unstable angina (USA). Both are invariably caused by recent thrombus formation on pre-existing coronary artery plaque leading to impaired myocardial oxygen supply. In this sense they differ from stable angina, which is usually precipitated by increased myocardial oxygen demand with severe background coronary artery narrowing. Both represent medical emergencies and are one of the most frequent causes of hospital and coronary care unit (CCU) admission.

AETIOLOGY AND RISK FACTORS
Atheroma deposits in the walls of coronary arteries provide the substrate for the development of ACS. Major risk factors for the development of coronary artery atheroma are seen in Figure 16.2. Up to 90% of the adult population possess at least one risk factor for the development of atheroma and CAD.
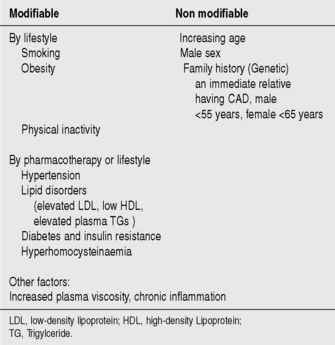
Cessation of smoking, lowering plasma cholesterol (diet and medications) and adoption of a more active lifestyle can all help prevent the development of CAD. Treatment of hypertensive patients produces a large and early reduction in stroke incidence (35–40%) and mortality, and a significant fall in MI (20–25%).3 Modification of risk factors is one of the most important means of decreasing the prevalence and mortality from CVD.
PATHOPHYSIOLOGY
Formation of thrombus upon disrupted, fissured or eroded atheromatous plaque is the usual precipitant of an ACS.4 Atherosclerotic plaque formation is probably initiated by injury to the vessel wall that may commence even as early as childhood. Highly activated macrophages are attracted to the site of injury and differentiate into tissue macrophages. Macrophages incorporate blood stream lipids into the connective tissue fibres of the plaque, forming a thrombogenic soft lipid core. Plaque development is slow, but is rapidly accelerated in people with risk factors.
‘Vulnerable plaque’ is often rich in lipid and covered by a thin fibrin cap. The cause of plaque rupture or fissuring is unknown but exposes thrombogenic lipid and collagen, which are potent activators of platelets. Development of thrombus upon this eroded plaque results from: (1) platelet adherence and activation; and (2) coagulation pathway activation.
Although many pathways initiate platelet activation, the final common pathway of thrombus formation is via activation of the glycoprotein (GP) IIb/IIIa receptor, the platelet surface membrane receptor for fibrinogen. Activated GP IIb/IIIa receptors cross-link fibrinogen between activated platelets, promoting the formation of platelet thrombi. Platelets aggregate to form ‘white thrombus’; however this thrombus is seldom totally occluding. Activation of coagulation pathways by exposed lipid and fibrin, as well as by the now activated platelets, leads ultimately to thrombin activation and the laying-down of fibrin clot. Red cells are enmeshed in this so-called ‘red thrombus’ complex, which surrounds the ‘white thrombus’. Sudden artery occlusion by thrombus may thus complicate even only moderate-sized plaque; 70% of ACS patients may have a < 50% stenotic lesion and in only 14% is the underlying stenosis > 70% of the lumen diameter (Figure 16.3).
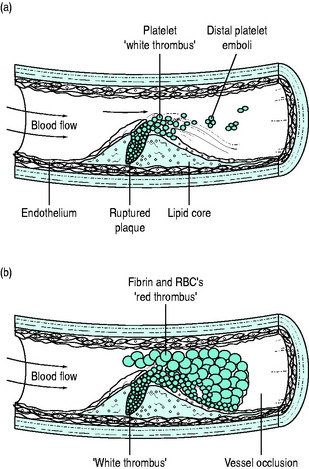
These processes have immediate relevance to treatment:
Totally occluding thrombus causes myocardial necrosis unless there is good collateral flow or the thrombus is rapidly cleared. Occlusion is often accompanied by ST-segment elevation on the ECG. If thrombus is largely ‘white thrombus’, with minimal or non-occlusive red thrombus, ST-segment elevation is far less likely. Non-occlusive thrombus may be asymptomatic, may cause USA or may cause MI, especially if spasm or distal embolisation of thrombus occurs. Although non-occlusive thrombus is less likely to be associated with early or sudden death, it is indicative of unstable plaque and is strongly associated with reinfarction and death in following months.
CLINICAL PRESENTATION
The diagnosis of myocardial ischaemia is usually made (suspected) on the basis of clinical history and ECG.2
HISTORY
Typically, the pain of acute MI:
Sweating, nausea, pallor, dyspnoea and anxiety are common.
The pain may sometimes be atypical:
These features do not necessarily exclude infarction.5 Differential diagnosis includes:
Atypical or silent presentations are common: 20–60% of non-fatal infarctions are unrecognised at onset.4 This presentation is more common in patients who are elderly, diabetic, have hypertension, who smoke or take non-steroidal anti-inflammatory agents.
PHYSICAL EXAMINATION
Examination of patients with USA is often unremarkable. With more severe infarction and extensive myocardial injury, signs of autonomic activation (pallor, sweating, agitation, clamminess) as well as heart failure and even shock may be apparent. Pericardial friction rubs occur frequently after MI but are usually transient.
Cardiogenic shock, hypotension, oliguria and other features of low cardiac output are associated with particularly poor outcome. Shock may be present without hypotension6 and may develop many hours after onset of symptoms. Right ventricle (RV) infarction results in hypotension and marked elevation of the jugular venous pressure (also seen with major LV dysfunction, usually in association with marked pulmonary venous congestion).
Conditions with similar presentations that do not benefit from thrombolysis are:
INVESTIGATIONS
The presence of MI should also be qualified by:2
Technological advances allow accurate detection of very small infarcts (myocardial necrosis < 1.0 g)7 that would not have been detected in earlier eras.
ELECTROCARDIOGRAPHY
Identification of classical acute and early changes where ST-segment elevation MI (STEMI) is present identifies patients in whom reperfusion therapy may interrupt, prevent or minimise myocardial necrosis (Figure 16.4). These are:
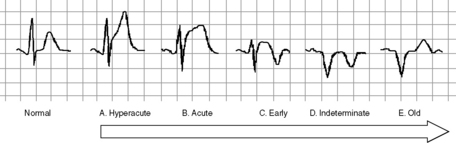
The Takotsubo syndrome is characterised by precordial ST-segment elevation, apical ballooning on echocardiography but normal vessels on angiography. It may follow the onset of recent severe stress and may cause up to 1–2% of STEMI.9 It has been recognised in critical illness.10
Patients with ACS but without significant ST-segment elevation (generically, non-ST-segment elevation ACS (NSTEACS), until further subdivided by biomarker studies) may still be at high risk of infarction and death. They likely have active, non-occluding thrombus or, if it is occluding, then some collateral flow is present. ECGs in these patients may be normal or display:
LOCALISATION OF INFARCTION
The left anterior descending (LAD) coronary artery supplies the anterior two-thirds of the interventricular septum (septal perforators), the anterior and lateral wall of the LV (diagonal branches) and sometimes part of the RV. The left circumflex artery supplies the LV lateral (anterolateral marginal branches) and posterior walls, and occasionally its inferior aspect (posterior LV arteries: 15% of patients) and the posterior septum. The right coronary artery (RCA) supplies the RV wall, and usually the posterior septum and inferior (diaphragmatic) wall of the left ventricle (posterior LV arteries; 85% of people). The RCA is ‘dominant’ (as opposed to the circumflex) if it gives rise to the posterior descending coronary artery (PDA) (Figure 16.5).
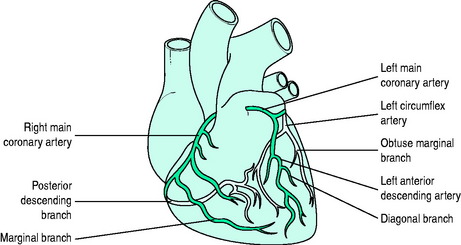
It is usual to use the ECG in initial clinical assessments to localise the area of myocardial ischaemia. The pattern of lead involvement may thus assist with localisation of the MI (Figure 16.6).8,11,12 There is a reasonable correlation between the site of infarction as defined by the ECG and the occluded coronary artery and the infarcted region of myocardium (Figure 16.7). However, ECG localisation may differ from angiographic, echocardiographic and autopsy findings, especially where there is collateral circulation or previous CABG. Anterior wall infarctions usually result from occlusion of the LAD; inferior, true posterior and RV infarctions result from occlusion of the RCA or circumflex arteries.
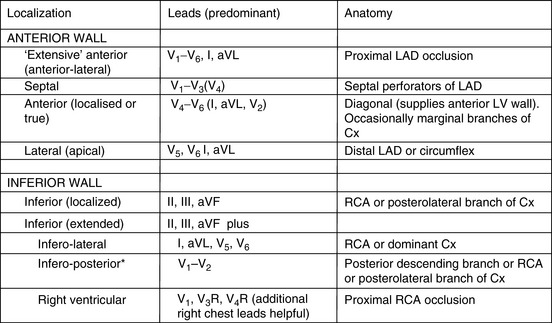
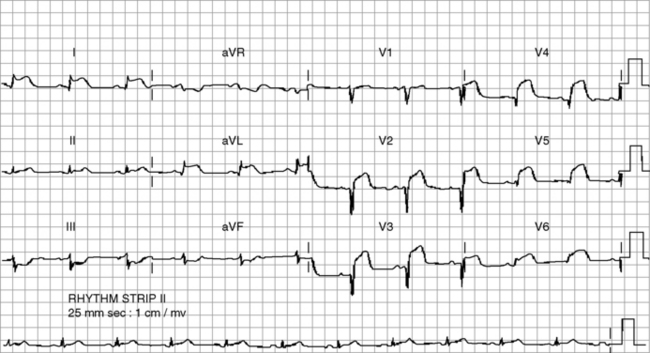
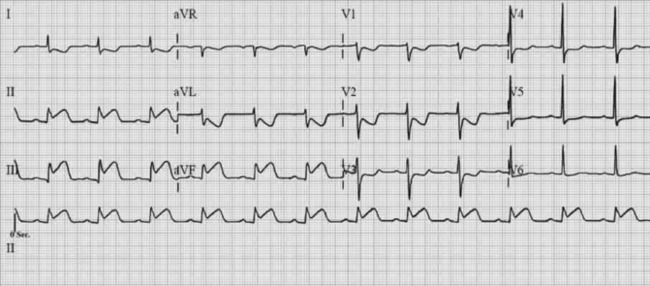
Common ECG patterns of infarction are shown in Figure 16.6. Approximately 40–50% of patients present with anterior infarction and 50% with inferior infarction. Anterior wall infarctions may be extensive or localised (septal, anterior, lateral) whereas inferior infarctions may similarly involve extension to the lateral, posterior or RV myocardium. Of clinical importance:
The resting ECG does not have sufficient predictive value to stratify patients with NSTEACS reliably into those with infarction (NSTEMI) and those without (USA). Up to 18% of patients with MI show no changes on the initial ECG and up to 20% of patients with NSTEACS have normal or minimal CAD.11,12 Cardiac biomarkers are necessary to confirm myocardial cellular injury and meet diagnostic criteria for MI.
CARDIAC BIOCHEMICAL MARKERS
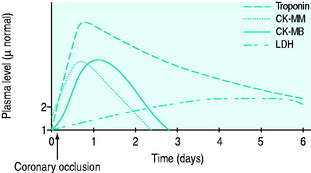
The typical rise and fall of cardiac markers after infarction are shown in Figure 16.8.
TROPONINS
The increased sensitivity of troponins compared to CK means that a third of patients previously considered to have USA are now recognised or redefined as having evidence of myocardial necrosis. Troponins also have better specificity than CK, similar to that of its isomer CK-MB. They do not differentiate the cause of the myocardial injury (e.g. ischaemia, myocarditis, trauma), however, and thus clinical context must always be considered.2 Troponins are more persistent in the serum (up to 7–10 days) and thus may be useful in diagnosis when presentation is delayed. Whilst CK-MB has traditionally been thought to be a better predictor of reinfarction, a rise in troponin of > 20% some 3–6 hours after onset of suspected reinfarction is also significant.2
Troponins should be checked in all patients with ACS and aggressive therapies should be targeted at patients with elevated levels.5
TROPONINS IN CRITICAL ILLNESS
Cardiac-specific troponins are frequently elevated in critical illness, including but not confined to sepsis, pulmonary embolus and renal insufficiency.13 It is likely that they are of cardiac origin and are indicative of myocardial injury. Their elevation does correspond with adverse outcomes. Most will not be due to ‘unstable plaque’, however, and the role of standard NSTEMI therapies is uncertain. Clinical context is necessary to decide those likely to have significant underlying CAD that may require acute or subsequent investigation.2
Please also see the latest information at the end of the chapter.
ECHOCARDIOGRAPHY
Echocardiography detects regional wall motion abnormalities, which can help confirm or exclude the diagnosis of MI in the small percentage of cases where diagnosis is uncertain (e.g. left bundle branch block (LBBB) or old infarction with atypical presentations). Regional wall motion abnormality and loss of wall thickening with contraction are often present in these cases if due to ischaemia, while their absence suggests that ischaemia is not acute. It is useful for excluding differential diagnoses (e.g. aortic dissection or pericardial effusions), again in a small percentage of patients.2
Echocardiography is subsequently useful to:
STRESS TESTING
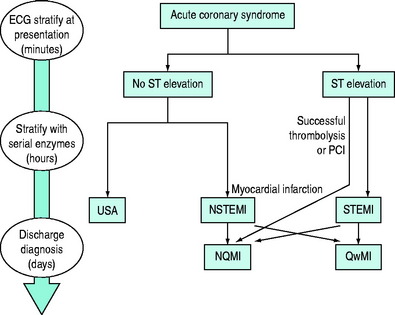
STEMI
STEMI (including new, presumed new LBBB) with persistent pain is the most lethal form of ACS and is usually due to complete occlusion of a coronary artery (> 90% of patients).5 It is an indication for reperfusion therapy. Such therapy may be successful and significantly reduce the size of the potential infarction, although some rise in troponin is usually inevitable. Patients who develop ST-segment elevation after admission should also then be stratified to this group.
NSTEACS
The therapy of NSTEMI aligns more clinically with that of USA than with that of STEMI. These two conditions, both forms of NSTEACS, represent a spectrum of disease and require common treatments directed at platelet inactivation and ‘plaque stabilisation’. The term ‘NSTEACS’ recognises that they are clinically indistinguishable at presentation. The more severe the ischaemia, the higher the need for more aggressive anticoagulation and invasive procedures. Early diagnostic classification using both ECG and troponins allows early risk stratification and evidence-based therapy. Only 35–75% of patients have evidence of coronary thrombus formation and thrombolytic therapy is not beneficial in this group. Indeed, it is associated with worse outcomes.5,14
Figure 16.10 displays the incidence of major coronary events over the following 12 months in patients presenting with and without ST-segment elevation. ST-segment depression has lesser early mortality but a similar or higher mortality at 6 months and at 10 years than those presenting with ST-segment elevation.15,16 Features that correlate with risk are:
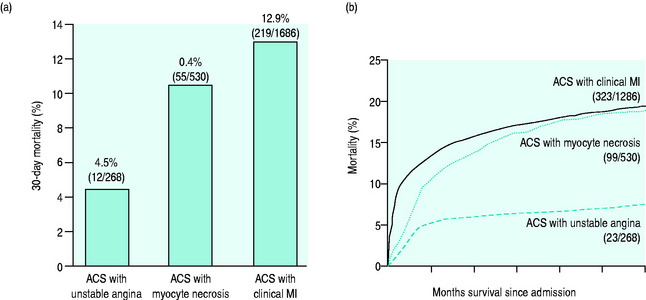
Q-wave MI (QwMI) or non-Q-wave MI (NqwMI) are older terms used to describe MI. The 10-year mortality of NqwMI (70%) is 10% higher than that of QwMI.16
IMMEDIATE MANAGEMENT OF ACUTE CORONARY SYNDROMES
PREHOSPITAL CARE
Review of trials comparing prehospital to in-hospital thrombolysis suggests a 17% decrease (95% confidence interval, 2–29%) in 30-day mortality.17


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


