INTRODUCTION
Normal regulation of bleeding is a complex process involving platelets and the coagulation system (see chapter 232, “Tests of Hemostasis”). Platelet defects typically manifest with petechiae and mucosal bleeding, whereas coagulation defects usually present as spontaneous or excessive hemorrhage. Management of significant acquired bleeding disorders should be discussed with a hematologist because there are often subtleties in diagnosis and treatment.
ACQUIRED PLATELET DEFECTS
Circulating platelets provide the initial defense against bleeding. Acquired platelet disorders may be quantitative (decreased number of circulating platelets) or qualitative (poorly functioning platelets). Both significant thrombocytopenia and qualitative platelet dysfunction are commonly manifested by the presence of nonpalpable petechiae, often most pronounced in the lower extremities and in areas where blood flow is restricted. Other typical findings may include purpura, mucosal bleeding (gingival, epistaxis), menorrhagia, hemoptysis, hematuria, and hematochezia, whereas deep tissue bleeding and hemarthrosis are less common.
Securing circulatory stability is the primary treatment priority in the bleeding patient. Once this is done, the history, physical examination, and directed laboratory testing are used to define the clinical syndrome. Recent illness, symptoms, and medications are potentially relevant in the patient who appears to have a platelet disorder. Physical examination should assess for additional bleeding sites and the type of bleeding. Assess for lymphadenopathy, splenomegaly, pallor, or jaundice that suggests diagnoses such as leukemia, lymphoma, systemic lupus erythematosus, infectious mononucleosis, or hemolytic anemia.
Quantitative defects that result in thrombocytopenia are caused by decreased production, increased destruction, splenic sequestration, platelet loss, or a combination (Table 233-1).1 The etiologies of thrombocytopenia are diverse, and the pathologic mechanism is not always initially clear. Viral infections may impair thrombopoiesis due to direct infection of the megakaryocyte, toxic effects of viral proteins or cytokines, hemophagocytosis, or production of antibodies that bind to platelets and enhance immune destruction.2 A drug history is important, because many medications have been implicated in causing thrombocytopenia or impairing platelet function (Table 233-2).3,4
| Mechanism | Associated Clinical Conditions |
|---|---|
| Decreased platelet production | Marrow infiltration (tumor or infection) Viral infections (rubella, HIV, hepatitis C, others) Drugs (Table 233-2) Radiation Vitamin B12 and/or folate deficiency |
| Increased platelet destruction | Immune thrombocytopenia Thrombotic thrombocytopenic purpura Hemolytic-uremic syndrome Disseminated intravascular coagulation Viral infections (HIV, mumps, varicella, Epstein-Barr virus) Drugs (Table 233-2) |
| Platelet loss | Excessive hemorrhage Hemodialysis, extracorporeal circulation |
| Splenic sequestration | Sickle cell disease, cirrhosis |
| Produce Thrombocytopenia | Impair Function |
|---|---|
| Heparin 4+ | Aspirin |
| Gold salts 4+ | Nonsteroidal anti-inflammatory drugs |
| Sulfa-containing antibiotics 4+ | Glycoprotein IIb–IIIa agents: ticlopidine and clopidogrel |
| Quinine and quinidine 4+ | |
| Ethanol (chronic use) 4+ | Penicillins and cephalosporins |
| Aspirin 3+ | Calcium channel blockers |
| Indomethacin 3+ | β-Adrenergic blockers: propranolol |
| Rifampin 2+ | Nitroglycerin |
| Abciximab and eptifibatide 2+ | Antihistamines |
| Thiazides and furosemide 2+ | Phenothiazines |
| Acyclovir 2+ | Cyclic antidepressants |
| Procainamide 2+ | |
| Digoxin 2+ | |
| Cimetidine and ranitidine 2+ | |
| Phenytoin and valproate 1+ | |
| Penicillins/cephalosporins 1+ |
A CBC will establish the presence and degree of thrombocytopenia and determine whether other hematologic cell lines are affected. A peripheral smear should be examined to assess platelet morphology and evaluate other cell lines. Rarely, in vitro platelet agglutination occurs, which leads to spuriously low platelet counts reported by automated cell counters (pseudothrombocytopenia) when collected in ethylenediaminetetraacetic acid–coated blood tubes; this can be detected by the presence of platelet clumping on a peripheral smear. In this case, a correct platelet count may be obtained by using citrated or heparin-anticoagulated blood tubes.
Testing for human immunodeficiency virus and hepatitis C virus is recommended in cases of isolated thrombocytopenia, as low platelets may be the only readily apparent manifestation in early infections.5 If the etiology of low platelets remains unclear, further laboratory testing to evaluate for disorders such as hemolysis and disseminated intravascular coagulation (DIC) may be needed. Although usually not necessary in instances of isolated thrombocytopenia, bone marrow biopsy may be obtained when malignancy or other bone marrow pathologies are of concern.
When circulating platelet levels decrease to below 10,000 to 20,000/mm3 (10 to 20 × 109/L), the risk of spontaneous bleeding becomes concerning, particularly for intracranial hemorrhage. Additional risk factors for bleeding include age, comorbid illness (i.e, renal disease, liver disease, connective tissue disease, peptic ulcer disease, hypertension), fall risk, and lifestyle activity. With the exception of a few specific disease processes, platelet transfusion in the nonbleeding patient should be considered when counts fall below 10,000/mm3 (10 ×109/L), or higher if other comorbid illnesses are present.6 The cause of the platelet deficiency may also influence the risk of bleeding. At a given platelet level, patients with immune thrombocytopenia (ITP) typically bleed less than patients with aplastic anemia, as the younger platelets present in ITP are more effective in hemostasis.7
ITP is an acquired immune-mediated disorder in which circulating platelets typically fall to very low levels.3,8 ITP results from antiplatelet antibodies that attack platelet surface glycoproteins such as glycoprotein IIb/IIIa, leading to peripheral platelet destruction. There is evidence that the same antibodies lead to impaired platelet production by megakaryocytes.9 Despite very low platelet counts, the circulating platelets are not functionally impaired.
Immune thrombocytopenia may be classified as primary (formerly known as idiopathic thrombocytopenic purpura) or secondary when associated with other medical conditions (autoimmune disorders, infections) or drug exposures.8 This distinction is of clinical importance, as management of the underlying disorder or discontinuation of the drug in cases of secondary ITP may lead to normalization of the platelet count.
Primary ITP may occur at any age, with equal prevalence between genders, with the exception of those age 30 to 60 years, in which women predominate. Primary ITP is classified by duration of illness: newly diagnosed (diagnosis to 3 months), persistent (3 months to 12 months), and chronic (>12 months).8 The majority of adults with primary ITP progress to chronic illness.
The most common presenting sign of ITP is a petechial rash (Figure 233-1A). Mild epistaxis, gingival bleeding (Figure 233-1B), and menorrhagia in women of childbearing age may also be seen. Except for petechiae and bruising, the patient should have a normal physical examination.
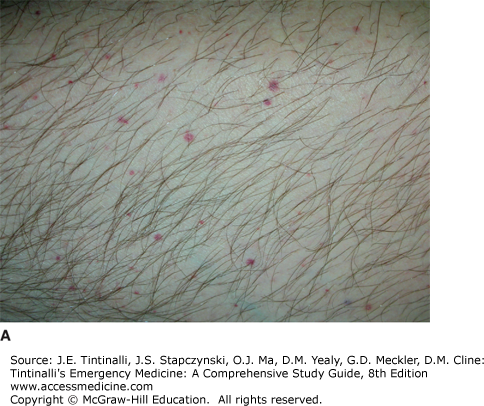
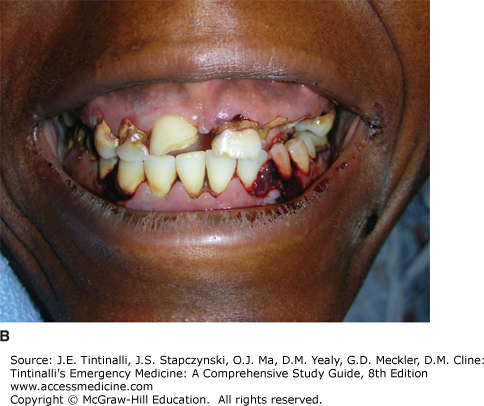
The CBC should demonstrate normal cell lines except for the platelets. A mild anemia may be seen if bleeding is present. The peripheral smear should show large, well-granulated platelets, although they will be few in number. Bone marrow biopsy is not indicated unless clinical features are atypical. If the evaluation supports the diagnosis of primary ITP, then additional testing in the ED is not required.
Treatment for ITP is initiated based on the risk of bleeding (Table 233-3). Major bleeding complications in patients with platelets >50,000 /mm3 (>50 × 109/L) are uncommon. As most fatal bleeding complications occur when platelet counts fall below 30,000/mm3 (30 × 109/L), experts and guidelines recommend this level as the threshold for initiating treatment of ITP in adults.5,6,9,10,11 Bleeding risk correlates with lower platelet counts and patient age greater than 60, a prior history of bleeding, certain chronic illnesses, and use of medications that alter hemostasis all heighten this risk. Therefore, certain patients may benefit from medical treatment despite a platelet count exceeding 30,000/mm3 (30 × 109/L). For all patients with primary ITP, bleeding risk should be minimized, including avoiding use of antiplatelet medications when possible, maintenance of good blood pressure control, optimized treatment of exacerbating comorbid conditions (i.e., liver disease, renal disease), and addressing fall risks.
| Platelet transfusion | <10,000–20,000/mm3 (<10–20 × 109/L) prophylactically to prevent bleeding <50,000/mm3 (<50 × 109/L) if actively bleeding <20,000–50,000/mm3 (<20–50 × 109/L) if patient requires diagnostic puncture <50,000–100,000/mm3 (<50–100 × 109/L) if patient requires invasive procedure |
| Corticosteroids | Prednisone, 1 milligram/kg PO per day for 21 days then tapered, or dexamethasone, 20 milligrams/m2 IV per day for 4 d |
| IV immunoglobulin | 1 gram/kg IV per day for 3 d |
| Splenectomy | Considered for refractory cases |
About two thirds of children with primary ITP will have spontaneous resolution within 6 months of diagnosis.11 An antecedent viral infection is commonly reported. Additionally, the measles-mumps-rubella vaccination has been associated with the development of acute ITP, although this is rare and occurs in less than 1 per 25,000 doses given.12 The risk of severe bleeding complications appears to be rare in children, regardless of their absolute platelet count.5,8,13 Children without signs of active bleeding and good psychosocial support typically do not require hospitalization or medical treatment. Good anticipatory guidance should be provided to the family, and the child should abstain from contact sports or activities that might place them at risk for head injury.
First-line treatment for primary ITP generally includes corticosteroids, typically prednisone 1 milligram/kg per day for 4 weeks (shorter courses may be considered for children).5,9,10,11,13 Initial response may not be seen for 4 to 14 days, and peak effect can be expected between 7 and 28 days. IV immunoglobulin may also be considered, either alone or in combination with corticosteroids, at a dose 1 g/kg/per day for 3 days. Initial response to IV immunoglobulin is more rapid than corticosteroids, occurring between 1 and 3 days, with peak effect seen between 2 and 7 days. Side effects of IV immunoglobulin may include aseptic meningitis, transient neutropenia, acute kidney injury, and transfusion reactions/hypotension.
Anti-Rho(D) immunoglobulin at 50 to 75 micrograms/kg for a single dose may also be considered as a first-line treatment strategy in patients who are Rh(D) positive and have not undergone splenectomy.14 Initial response rate and peak effect are similar to that seen with IV immunoglobulin. Mild hemolysis can be expected, and rare serious or even fatal cases of intravascular hemolysis, DIC, and renal failure have been reported with anti-Rho(D) use, prompting the U.S. Food and Drug Administration to administer a warning pertaining to its use in 2010. Second-line treatment modalities include splenectomy and use of newer agents, such as the monoclonal antibody rituximab, which blunts the autoimmune destruction of platelets, and thrombopoietin receptor agonists, which stimulate platelet production (i.e., romiplostin, eltrombopag).
In the event of life-threatening hemorrhage where hemostasis must be achieved rapidly, IV corticosteroids (such as dexamethasone) and IV immunoglobulin should be administered concomitantly.13 Platelet transfusions should be initiated, and doses two to three times the typical dose might be required due to persistent autoimmune destruction.6 Platelet transfusions may need to be repeated frequently until bleeding subsides or targeted treatment modalities take effect. Emergent splenectomy may be considered if all other treatment options are exhausted.
Over 200 drugs have been implicated in causing immune-mediated suppression of platelet production or increased platelet destruction (Table 233-2).3,4
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


