By definition, FENa is the ratio between the quantity of Na excreted in the urine relative to the amount filtered at the glomerulus. Measuring urine sodium concentration alone is not sufficient, as the sodium concentration in urine varies with water reabsorption. It is necessary to plug in the serum and urinary creatinine into the calculation, in order to calculate the amount of fluid and sodium that is filtered through to glomerulus [1, 2].
Prerenal AKI can be due to intrarenal vasoconstriction, systemic vasodilation, and volume depletion. These patients will try to compensate and retain sodium and usually have a FENa of less than 1%. If any of the above insults continue and become intense, the blood supply to the renal tubules is severely reduced leading to acute tubular necrosis. Once the tubules are damaged, they lose their ability to reabsorb sodium, and the FENa will usually be greater than 2–3%.
FENa is often used in the setting of acute renal failure to help distinguish between prerenal (decreased renal perfusion) and intrinsic renal (ATN due to renal hypoperfusion) causes. In general, a FENa of <1% suggests prerenal disease, between 1 and 2% is indeterminate, and >2% suggests ATN. There are some exceptions to this, but overall, the specificity of this test is >80%.
There are limitations to FENa. The threshold used to distinguish prerenal and intrinsic renal disease may vary; there are other causes of low FENa and salt-wasting conditions (like diuresis) affect urinary sodium levels.
- 4.
Contrast-induced nephropathy (CIN) is either a relative increase in serum creatinine from baseline value by 25% or an absolute increase of 0.5 mg/dL within 48 to 72 h after contrast exposure not attributable to other causes and must persist for 2 to 5 days. FENa may vary widely and in the minority of patients with oliguric CIN, the FENa may be low despite lack of clinical evidence of volume depletion [3].
Risk factors include pre-existing renal dysfunction, diabetes with renal dysfunction, age >70 years, cardiorespiratory disease, hypotension or dehydration, and nephrotoxic medications (NSAIDs or aminoglycosides). Contrast agent volume, route of administration (intra-arterial), hyperosmolarity, and multiple doses in 72 h also add to the risk.
Measures to decrease risk of CIN include prehydration with saline, using the lowest dose of low osmolar contrast, IV bicarbonate infusion, N-acetylcysteine (controversial), discontinuation of nephrotoxic drugs for 48 h prior to contrast, and the use of hemofiltration (expensive) pre- and post-contrast use.
- 5.
Fractional excretion of other substances such as urea and uric acid can also be measured to determine their renal clearance to help distinguish prerenal from intrinsic renal causes. The FEUrea may be more accurate in distinguishing ATN from prerenal disease in patients being treated with diuretics since diuretics as mentioned earlier cause natriuresis.
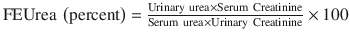
- 6.
The differential diagnosis of acute kidney injury (AKI) or acute renal failure (ARF) in this patient with cirrhosis includes prerenal azotemia, acute tubular necrosis, and hepatorenal syndrome (HRS). Prerenal azotemia is caused by hypovolemia (e.g., aggressive diuresis, diarrhea, and/or gastrointestinal bleeding) or by other causes of decreased effective blood volume induced by infections or vasodilators. Prerenal azotemia responds to volume expansion, and vasoconstrictors and dialysis are not required.
Acute tubular necrosis mostly occurs in patients presenting with shock or a history of exposure to nephrotoxins/contrast agents. Acute tubular necrosis is treated with renal replacement therapy if indicated.
HRS occurs in patients with cirrhosis or liver failure when there is a sudden rapid deterioration of liver function due to an insult like gastrointestinal bleed, infection, or excessive diuresis. It is caused by extreme vasodilatation with consequent renal vasoconstriction and is treated with vasoconstrictors and volume expansion with albumin. HRS remains a diagnosis of exclusion. Therefore, the first step in its diagnosis is to exclude the presence of structural kidney injury (acute tubular necrosis, glomerulonephritis, and acute interstitial nephritis) or obstructive kidney injury (obstructive uropathy) and to then distinguish between prerenal azotemia and HRS (the two functional types of AKI in cirrhosis).
- 7.
Renal dysfunction in HRS is functional. The pathophysiology of cirrhosis involves portal hypertension leading to splanchnic arterial vasodilatation. The resultant primary systemic arterial vasodilatation leads to systemic hypotension which in turn causes activation of the neurohumoral axis with stimulation of the renin–angiotensin–aldosterone system (RAAS), sympathetic nervous system (SNS), and arginine vasopressin (AVP). Stimulation of the RAAS, SNS, and AVP contributes to maintenance of blood pressure by increasing systemic vascular resistance along with the secondary increase in cardiac output. While this compensatory neurohumoral activation attenuates any hypotension secondary to arterial vasodilatation, renal vasoconstriction with sodium and water retention also occurs. This resultant diminished renal function is, however, of a functional nature and thus should not be considered ATN in the initial phases. Prolonged and severe HRS can then lead to ATN [4, 5].Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree





