Toxicologic
24-1 Gastrointestinal Decontamination
Anthony Morocco
The appropriate use of gastrointestinal decontamination after potentially harmful ingestions remains controversial.1,2,3,4,5 Modalities include single- and multi-dose activated charcoal (AC), gastric lavage, cathartics, and whole-bowel irrigation (WBI). All of these techniques have theoretical value in limiting absorption of poisons, but little or no evidence exists for improved outcomes.
Ipecac is an emetic agent used for gastric emptying. This drug is now rarely used, because its administration can cause prolonged vomiting, aspiration, and esophageal injury.1
Orogastric lavage involves the use of a large tube (32F to 40F in adults or 22F to 24F in children) in an attempt to retrieve intact pills or pill fragments from the stomach. The stomach is flushed with aliquots of water until a clear effluent is obtained. Potential complications include aspiration and injury to the oropharynx, esophagus, or stomach.2
AC adsorbs most chemicals in the gut and prevents absorption. This is considered an important treatment for most ingestions. Multi-dose AC involves repeated doses given every 2 to 4 hours. AC administration may result in vomiting and aspiration or bowel obstruction.3,4 One dose of a cathartic, such as sorbitol, may speed the movement of substances through the gastrointestinal tract, but sorbitol should not be used alone or in multiple doses.
WBI uses polyethylene glycol electrolyte solution (PEG-ES) to cleanse the bowel of toxic agents before they are absorbed. The dose is 1 to 2 L/hour via nasogastric tube in adults, 1 L/hour in children ages 6 to 12 years, and 500 mL/hour in children ages 9 months to 6 years, given until rectal effluent is clear. WBI is used in the setting of ingestion of enteric-coated or sustained-release preparations. Nausea, vomiting, and abdominal pain may occur.5
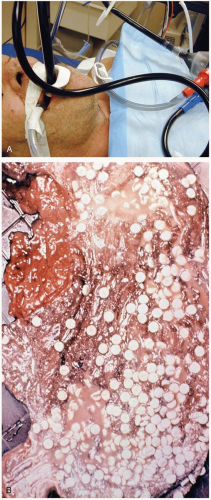 FIGURE 24-1 A: Gastric lavage. (Courtesy of Robert Hendrickson, MD.) B: Gross autopsy specimen of stomach with multiple pills adherent to the gastric wall. (Copyright James R. Roberts, MD.) |
REFERENCES
1. Krenzelok EP, McGuigan M, Lheur P. Position statement: ipecac syrup. J Toxicol Clin Toxicol 1997;35:699-709.
2. Vale JA. Position statement: gastric lavage. J Toxicol Clin Toxicol 1997;35:711-719.
3. Chyka PA, Seger D. Position statement: single-dose activated charcoal. J Toxicol Clin Toxicol 1997;35:721-741.
4. Chyka PA, Seger D. Position statement and practice guidelines on the use of multi-dose activated charcoal in the treatment of acute poisoning. J Toxicol Clin Toxicol 1999;37:731-751.
5. Tenenbein M. Position statement: whole bowel irrigation. J Toxicol Clin Toxicol 1997;35:753-762.
24-2 Caustic Ingestion: Alkali
Gregory Schneider
Clinical Presentation
Signs of caustic alkali ingestion include injury to the lips, oral mucosa, tongue, and pharynx. With intentional ingestions, if liquids are gulped quickly, there may be relative sparing of the oral and pharyngeal mucosa.1
Pathophysiology
Alkali substances cause injury by liquefaction necrosis. Ingestion of a substance with a pH of 12 or greater often involves drain cleaners, other cleansers, household bleach, Clinitest tablets, or button batteries. The extent of the injury is determined by four factors: the pH of the material, the amount ingested, solid versus liquid form, and the duration of exposure. In the first 24 to 48 hours, tissue damage is aggravated by bacterial invasion. Strictures may result 4 to 6 weeks after exposure.2
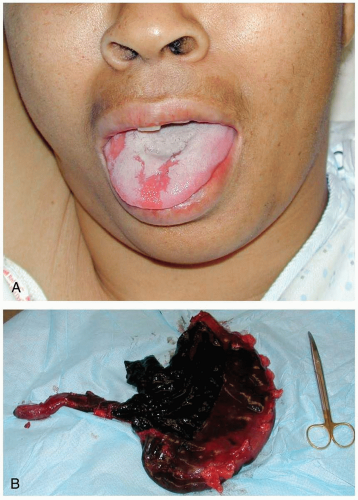 FIGURE 24-2 A: Alkaline burn of the tongue after ingestion of sodium hydroxide. (Courtesy of Robert Hendrickson, MD.) B: Extensive necrosis of gastrointestinal tract after ingestion of caustic liquid. (Courtesy of William Hughes, MD, and Laura Spivak, MD.) |
Diagnosis
Clinical Complications
Extensive mucosal damage leads to fever, tachypnea, tachycardia, hypotension, and shock. Perforations of the esophagus and stomach, leading to mediastinal or peritoneal abscess, sepsis, and death, have occurred. Aspiration pneumonia, burns of the epiglottis and vocal cords, and laryngeal obstruction may also occur. Long-term sequelae include esophageal stricture, vocal cord paralysis, and pyloric stenosis.3
Management
Dilution with milk or water should be initiated rapidly. Attempts to prevent vomiting are important to avoid reintroduction of the substance into the esophagus and oropharynx. Asymptomatic patients should be observed for 4 to 6 hours. Patients ingesting bleach have a low incidence of serious injury. Patients with refusal to swallow, nausea, vomiting, abdominal pain, drooling, coughing, dysphagia, stridor, or oral burns should be admitted to the hospital for possible endoscopy and observation.3 Endoscopy should occur within 24 hours to determine the extent of injury and subsequent treatment. Steroid treatment remains controversial and should not be initiated until the extent of the injury is determined.1
REFERENCES
1. Wason S. The emergency management of caustic ingestions. J Emerg Med 1985;2:175-182.
2. de Jong AL, Macdonald R, Ein S, et al. Corrosive esophagitis in children: a 30-year review. Int J Pediatr Otorhinolaryngol 2001;57:203-211.
3. Gorman RL, Khin-Maung-Gyi MT, Klein-Schwartz W, et al. Initial symptoms as predictors of esophageal injury in alkaline corrosive ingestions. Am J Emerg Med 1992;10:189-194.
24-3 Caustic Ingestion: Acid
Gregory Schneider
Clinical Presentation
Patients with caustic acid ingestion complain of pain in the oropharynx, esophagus, and abdomen. Other early signs include drooling, excessive salivation, and refusal to swallow. Burns may be seen on the lips, skin, and oropharynx. The intent of the injury must be determined, because suicidal and intentional ingestions are more commonly associated with serious injury.1
Pathophysiology
Acids produce coagulation necrosis that tends to limit the extent of the injury by preventing penetration of the acid into the tissue. Compared with alkali ingestions, acids cause less damage to the esophagus and more damage to the stomach and small intestine.1 Injury severity is determined by the pH and quantity of the substance, as well as the amount of time spent in the stomach. Pyloric spasm, antral mucosal edema, inflammation, and, ultimately, pyloric stricture may occur after acid ingestion.2 Peritonitis and intestinal perforation are not uncommon.1
Diagnosis
Once it is determined that a corrosive ingestion has occurred, it becomes urgent to determine the extent of the injury. A complete blood count, electrolyte panel (including calcium, magnesium, and phosphate levels), upright chest radiograph (looking for perforation), arterial blood gas determination, and electrocardiography should all be performed.1,2,3
Clinical Complications
Management
Dilution with water or milk should be initiated immediately. If perforation is suspected, a laparotomy should be performed to determine the viability of the stomach. Stable patients should be evaluated endoscopically. Hydrofluoric acid may bind calcium and magnesium, leading to serious electrolyte disorders. Aggressive repletion of calcium and magnesium should be initiated empirically.3
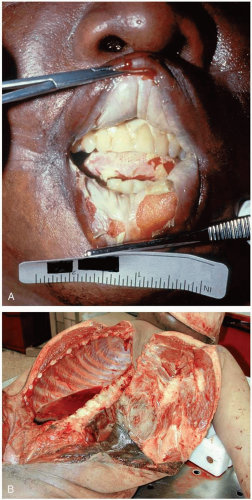 FIGURE 24-3 A: Postmortem oral burns in a patient who drank battery acid. B: Postmortem internal abdominal wall burns in a patient who drank battery acid. (A and B, Courtesy of Mark Su, MD.) |
REFERENCES
1. Wason S. The emergency management of caustic ingestions. J Emerg Med 1985;2:175-182.
2. Gun F, Abbasoglu L, Celik A. Acute gastric perforation after acid ingestion. J Pediatr Gastroentrol Nutr 2002;35:360-362.
3. Yu-Jang S, Li-Hua L, Wai-Mau C, et al. Survival after a massive hydrofluoric acid ingestion with ECG changes. Am J Emerg Med 2001;19:458-460.
24-4 Body Stuffing
Anthony Morocco
Clinical Presentation
The term “body stuffing” refers to the practice of hastily ingesting illicit drugs in order to dispose of evidence. In contrast to body packers, who swallow carefully packaged drugs for smuggling, body stuffers may swallow substances that are unwrapped or loosely wrapped in plastic, balloons, paper, or aluminum foil.1,2 Consequently, these patients are at high risk for drug absorption and subsequent toxicity.
Patients typically ingest the drug when confronted by police, who subsequently bring suspects to the hospital for treatment. Patients often deny ingestion or give an inaccurate history concerning the amount ingested. Most patients are asymptomatic. If the body stuffing goes undetected by police, symptoms may occur while the patient is in jail. These symptoms may be unexplained at the time of presentation. With cocaine and other sympathomimetic agents, agitation, tachycardia, seizures, and ventricular dysrhythmias occur. Heroin results in decreased level of consciousness, miosis, and hypoventilation.
Diagnosis
The diagnosis can be difficult due to an inaccurate or absent history. Drug ingestion should be strongly suspected in any patient who is brought from jail with loss of consciousness, seizure, agitation, or cardiopulmonary arrest. Plain radiography is unlikely to show drug packets in small radiolucent packaging. Contrast-enhanced studies may be more helpful. A positive urine immunoassay drug screen may signify recent exposure to the drug but is not useful in confirming or ruling out drug-packet ingestion.
Clinical Complications
Patients may develop rapid onset of seizures, coma, and death. Symptoms may be delayed for hours after ingestion due to delayed liberation of the drug from its packaging.
Management
Patients should be given oral activated charcoal and placed on a cardiac monitor. Further management is controversial, because most patients remain asymptomatic.1 Given the possibility of delayed toxicity and retained drug packets in the gut, whole-bowel irrigation with polyethylene glycol and admission to the intensive care unit for 24 hours of observation should be considered. Patients with symptoms of heroin ingestion should be treated with naloxone infusion. Cocaine toxicity is treated with benzodiazepines for sympathomimetic symptoms, vasodilators such as nitroprusside or phentolamine for hypertension, and, potentially, surgical retrieval of packages.1,2
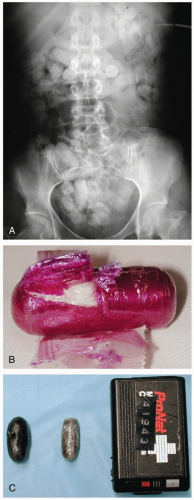 FIGURE 24-4 A: Abdominal radiograph revealing multiple packets containing cocaine. (Courtesy of Anthony Morocco, MD.) B: Drug packet containing cocaine. (Courtesy of Anthony Morocco, MD.) C: Packets containing hash. (Courtesy of Robert Hendrickson, MD.) |
REFERENCES
1. June R, Aks SE, Keys N, et al. Medical outcome of cocaine bodystuffers. J Emerg Med 2000;18:221-224.
2. Sporer KA, Firestone J. Clinical course of crack cocaine body stuffers. Ann Emerg Med 1997;29:596-601.
24-5 Smoke Inhalation
Anthony Morocco
Clinical Presentation
Patients with smoke exposure may present with symptoms of mucous membrane and upper airway irritation, including burning eyes, cough, and shortness of breath, as well as signs and symptoms of lower respiratory tract injury, including dyspnea, chest pain, and hemoptysis. In severe cases, they may present with respiratory distress or apnea, loss of consciousness, and, in some cases, concomitant cutaneous burns. Hallmarks of inhalation injury include the production of soot-stained sputum and singed nasal or facial hair.1
Pathophysiology
Smoke inhalation is the primary cause of death for fire victims. Smoke contains a variety of chemicals, depending on the burning material. These include carbon monoxide, carbon dioxide, acrolein, cyanide, phosgene, sulfur dioxide, nitrogen oxides, particulates (soot), and other substances. Patients who sustain cutaneous burns with concomitant smoke-related injury are more likely to die.1,2
Smoke exposure may result in respiratory system dysfunction by direct thermal injury, neutrophil-mediated production of free radicals and inflammatory mediators, destruction of lung surfactant, bronchospasm, airway edema, bronchorrhea, and ciliary dysfunction. Harmful chemical gases act by causing asphyxiation, systemic toxicity, or direct lung injury.1
Diagnosis
Injury from smoke inhalation should be suspected in any patient who has been exposed to a fire in a closed space, particularly if the patient exhibits respiratory or airwayrelated symptoms, singed facial or nasal hair, or carbonaceous sputum. Chest radiographic findings may include patchy atelectasis and global interstitial and alveolar infiltrates. These developments may be delayed for 24 to 36 hours. Bronchoscopy, pulmonary function tests, and ventilation-perfusion scanning may help to assess the extent of lung injury. Venous blood should be obtained for carboxyhemoglobin and cyanide concentrations, and arterial blood gases may need to be assessed frequently.1
Clinical Complications
Acute complications include death, severe lung injury, acute respiratory distress syndrome, pneumonia, and various thermal injuries.1 Chronic lung sequelae include reactive airway disease, bronchitis, and fibrosis.
Management
Most patients with smoke inhalation require hospitalization. Initial treatment is directed to the maintenance of a patent airway. Early endotracheal intubation should be considered for patients with potential airway injury, as evidenced by severe or progressive respiratory difficulties, significant facial or neck burns, or upper airway
edema. Supportive care should include humidified oxygen, aggressive fluid resuscitation, bronchodilators, and pulmonary toilet. Prophylactic antibiotics and steroids are not indicated. Specific treatments for carbon monoxide (hyperbaric oxygen) and cyanide (nitrites, thiosulfate) may be required.1
edema. Supportive care should include humidified oxygen, aggressive fluid resuscitation, bronchodilators, and pulmonary toilet. Prophylactic antibiotics and steroids are not indicated. Specific treatments for carbon monoxide (hyperbaric oxygen) and cyanide (nitrites, thiosulfate) may be required.1
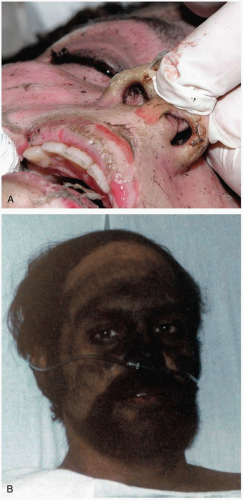 FIGURE 24-5 A: Singed nasal hairs. (© David Effron, MD, 2004. Used with permission.) B: Soot-stained face. (Courtesy of Michael Greenberg, MD.) |
REFERENCES
1. Lee-Chiong TL Jr. Smoke inhalation injury. Postgrad Med 1999;105:55-62.
2. Nguyen, TT, Gilpin DA, Meyer NA, et al. Current treatment of severely burned patients. Ann Surg 1996;223:14-25.
24-6 Inhalant Abuse
Anthony Morocco
Clinical Presentation
Acute inhalant intoxication may appear to be similar to ethanol inebriation, with ataxia, slurred speech, and nystagmus. Chronic users may exhibit nonspecific symptoms such as headaches, dizziness, loss of appetite, and cough. Emergency care may be sought only after sudden loss of consciousness and cardiac arrest.1
Pathophysiology
Inhalant abuse involves the breathing of volatile substances in order to experience a euphoric effect. Methods of inhalation include breathing a product directly from its container or a surface (“sniffing” or “snorting”), from a soaked rag placed over the face (“huffing”), or from a bag filled with a volatile substance (“bagging”). Twenty percent of adolescents report having tried inhalants by eighth grade.1 Commonly abused substances include paint thinner, airplane glue, spray paint, lighter fluid, hair spray, correction fluid, and nail polish remover. Specific substances include toluene, butane, propane, fluorocarbons, chlorinated hydrocarbons, and acetone.1
The mechanism of action of inhalants is unknown, but they are believed to act in the central nervous system in a manner similar to volatile anesthetic agents. Hydrocarbons are known to sensitize the myocardium to the arrhythmogenic effects of catecholamines.
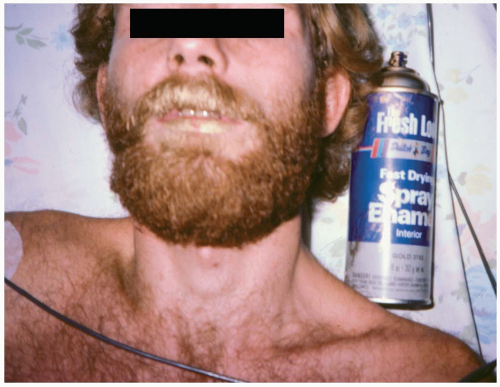 FIGURE 24-6 Gold paint on the mouth and mustache of a patient who died after huffing. (Copyright James R. Roberts, MD.) |
Diagnosis
Diagnosis is based on history and may be aided by noting a chemical odor or paint stains on the patient. Inhalant abuse should be suspected in any patient, particularly an adolescent, with nonspecific complaints, neurologic dysfunction, or sudden cardiac arrest.
Clinical Complications
Sudden sniffing death syndrome may occur during inhalant use if the person is startled while intoxicated, causing a catecholamine surge that triggers a ventricular arrhythmia. Other potential complications include aspiration, accidental trauma, respiratory depression, vagal inhibition, injuries or burns due to explosion, and cold injuries. Breathing a pure inhalant gas without air can result in anoxic death. Chronic use can cause myocarditis, myocardial fibrosis, bone marrow suppression, and fetal solvent syndrome. Chronic neurotoxicity includes peripheral neuropathy, cerebellar dysfunction, and dementia, with atrophy evident on brain imaging. A withdrawal syndrome may also occur. Toluene abuse can cause lung injury, renal tubular acidosis, and renal failure.1 Other agents with unique toxicities include methylene chloride (metabolized to carbon monoxide), carbon tetrachloride (hepatotoxicity), and nitrites (methemoglobinemia).2
Management
REFERENCES
1. Anderson CE, Loomis GA. Recognition and prevention of inhalant abuse. Am Fam Physician 2003;68:869-874.
2. Lorenc JD. Inhalant abuse in the pediatric population: a persistent challenge. Curr Opin Pediatr 2003;15:204-209.
24-7 Hydrocarbon Pneumonitis
Anthony Morocco
Clinical Presentation
Patients with hydrocarbon pneumonitis may present with a strong odor of hydrocarbon, particularly with substances such as pine oil or gasoline. Gastrointestinal irritation with nausea, vomiting, abdominal pain, and throat pain are common. Aromatic or halogenated hydrocarbons, camphor, phenol, and pine oil may produce systemic symptoms such as central nervous system depression with ataxia, coma, seizures, and cardiac dysrhythmias. Low-viscosity hydrocarbons (e.g., gasoline, kerosene, turpentine) and the aromatic and halogenated hydrocarbons may produce respiratory symptoms that include shortness of breath, cough, and wheezing.1
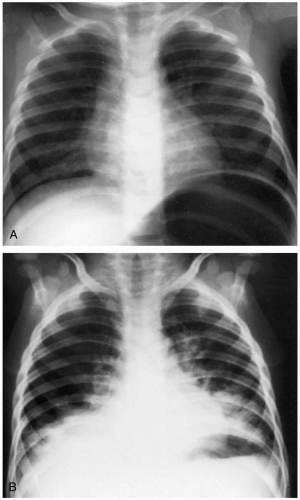 FIGURE 24-7 A: This patient aspirated red furniture polish; 2 hours after ingestion, the lungs are clear. B: By 12 hours after ingestion, extensive infiltrates in the lung bases are visible. (A and B, From Swischuk, with permission.) |
Pathophysiology
The mechanisms of hydrocarbon effects on various body systems are unknown. Dysrhythmias may occur after ingestion of halogenated hydrocarbons, due to myocardial sensitization to catecholamines. Lung injury most likely is caused by direct cellular injury and disruption of surfactant after aspiration. Three important determinants of risk for lung injury are viscosity, surface tension, and volatility of the hydrocarbon. Lower viscosity and lower surface tension increase the risk of aspiration, whereas higher volatility increases the volume of inhalation.1
Diagnosis
Diagnosis is based on the history, because no confirmatory laboratory studies are readily available. Patients with aspiration may give a history of coughing, choking, or gagging after ingestion. The odor of hydrocarbon on the patient helps confirm the diagnosis. Acute lung injury may be seen on chest radiographs as diffuse infiltrates. The radiographic findings usually develop within 6 hours, but may develop up to 24 hours after hydrocarbon exposure.1
Clinical Complications
Lung injury may progress to acute respiratory distress syndrome, respiratory failure, and death. Sequelae may include pulmonary fibrosis and bronchiectasis.
Management
Activated charcoal should be administered only in cases of ingestion of a hydrocarbon that can have systemic toxicity. Gastric emptying by nasogastric or orogastric lavage may be considered after large ingestions, but these procedures may stimulate vomiting and lead to aspiration. Aggressive supportive care, including vasopressors and mechanical ventilation, may be necessary. Lung injury usually peaks at 72 hours after ingestion.1
REFERENCES
1. Welker JA, Zaloga GP. Pine oil ingestion: a common cause of poisoning. Chest 1999;116:1822-1826.
24-8 Acetaminophen
Anthony Morocco
Clinical Presentation
After acute acetaminophen, N-acetyl-p-aminophenol (APAP) overdose, patients may exhibit only mild, nonspecific symptoms such as nausea and vomiting. Diminished level of consciousness and shock may occur rarely after massive ingestions. Right upper quadrant abdominal pain and jaundice may appear 3 to 5 days after ingestion.1
Pathophysiology
APAP is an analgesic found in numerous over-the-counter preparations. It is the most common ingested agent in suicide attempts, and it is a common cause of fulminant hepatic failure in the United States. Unintentional overdose accounts for up to one third of APAP toxicity cases. The minimum toxic dose of APAP is considered to be at least 150 mg/kg.1
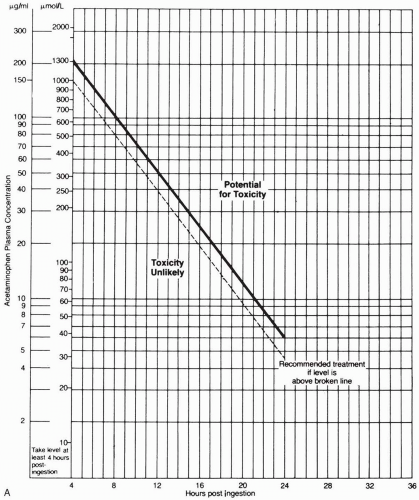 FIGURE 24-8 A: The modified Matthew-Rumack acetaminophen nomogram. (From Rumack, with permission.) |
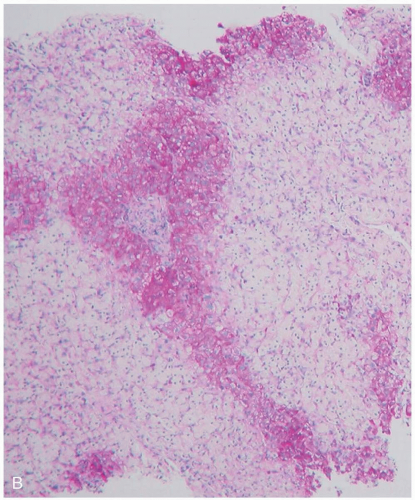 FIGURE 24-8 (cont’d) B: Centrilobular necrosis. (From Yamada, with permission.) |
APAP toxicity occurs because of excessive formation by liver cytochrome P450 of a specific metabolite, Nacetyl-p-benzoquinoneimine (NAPQI). With therapeutic APAP doses, only about 5% of the administered agent is converted to NAPQI, and this fraction is quickly detoxified by glutathione and other mechanisms. However, in APAP overdose, nontoxic metabolic pathways become saturated, and more NAPQI is produced than can be bound by glutathione. NAPQI then binds and disrupts various cellular proteins, resulting in cell death and zone 3 (centrilobular) hepatic necrosis. Toxicity is more likely in patients with decreased glutathione stores (fasting, alcoholism) or increased cytochrome P450 activity (alcoholism, isoniazid).1
Diagnosis
APAP ingestion should be suspected in any patient with acute liver failure. All patients with a suspected oral overdose should have a serum APAP concentration measured, regardless of their history.
Clinical Complications
Without treatment, hepatic injury may be self-limited, or it may progress to fulminant hepatic necrosis, resulting in coagulopathy, hepatic encephalopathy, and death. In addition, renal failure and myocardial injury may be seen.
Management
Oral activated charcoal should be administered if it is possible to do so within 1 to 2 hours after ingestion. The APAP concentration measured 4 to 24 hours after ingestion should be plotted on the Matthew-Rumack nomogram to determine the treatment course. If the concentration is above the line, signifying possible toxicity (150 µg/mL at 4 hours after ingestion), treatment with Nacetylcysteine (NAC) should be instituted. NAC is usually administered orally in the United States, though many U.S. institutions and those overseas give the drug intravenously. Patients with fulminant hepatic failure should be evaluated for possible liver transplantation.1
REFERENCES
1. Zimmerman HJ. Acetaminophen hepatotoxicity. Clin Liver Dis 1998;2:523-544.
24-9 Salicylate
Anthony Morocco
Clinical Presentation
Patients with acute salicylate overdose initially develop nausea and vomiting. Tinnitus is common and progresses to decreased auditory acuity. Worsening central nervous system toxicity causes agitation, delirium, decreased level of consciousness, and seizures. Hyperthermia occurs with severe toxicity. Patients with chronic toxicity are predominantly elderly and have an insidious onset of symptoms similar to those seen in acute overdose, such as nausea, vomiting, confusion, slurred speech, and delirium.1
Pathophysiology
Aspirin (acetylsalicylic acid, ASA) and related salicylates (bismuth subsalicylate, methyl salicylate) can cause severe poisoning after acute or chronic overdose. These compounds are found in a wide variety of oral and topical products. Of particular concern are enteric-coated ASA preparations, which can cause a significant delay in absorption and peak blood concentrations, and oil of wintergreen, which may have a very high salicylate concentration (equivalent to 1.4 g of ASA per milliliter).1
Salicylates cause hyperventilation and respiratory alkalosis by stimulation of the brainstem respiratory center. Anion gap metabolic acidosis occurs from a number of mechanisms, including uncoupling of oxidative phosphorylation and ketosis secondary to increased fatty acid metabolism. Because salicylate is a weak acid, systemic acidosis increases the amount of salicylate in nonionized form. This results in enhanced entry of salicylate into the central nervous system and worsening toxicity.1
Diagnosis
The diagnosis is confirmed by measurement of the serum salicylate concentration. Addition of ferric chloride to urine produces a deep purple color if salicylate is present.
Clinical Complications
Noncardiogenic pulmonary edema may occur after overdose. Death occurs from cerebral edema and cardiovascular collapse.1 Reye’s syndrome is a liver disorder associated with salicylate use in children.
Management
Patients should receive multidose activated charcoal to decrease absorption and enhance elimination of salicylate. Salicylate concentration and pH should be frequently measured. Whole-bowel irrigation may decrease absorption after ingestion of an enteric-coated preparation. Bicarbonate should be administered to alkalinize the serum and urine, thereby decreasing central nervous system entry and enhancing elimination in the urine by ion trapping. Hemodialysis is indicated for severe toxicity.1
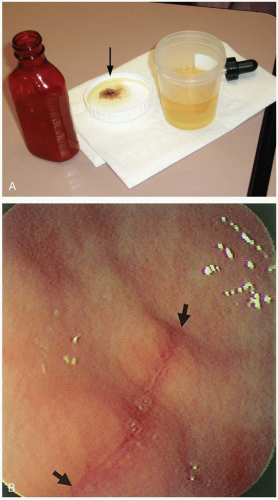 FIGURE 24-9 A: A positive ferric chloride test showing purple-brown color change in salicylate-containing urine when several drops of ferric chloride are added to a few milliliters of urine. (Courtesy of Judith Eisenberg, MD.) B: Characteristic of aspirin-induced linear erosion (extending between two arrows). (From Yamada, with permission.) |
REFERENCES
1. Dargan PI, Wallace CI, Jones AL. An evidence based flowchart to guide the management of acute salicylate (aspirin) overdose. Emerg Med J 2002;19:206-210.
24-10 Carbon Monoxide
Anthony Morocco
Clinical Presentation
Patients with carbon monoxide (CO) exposure may complain of a variety of nonspecific symptoms that may be mistaken for a flu-like illness. Headache is one of the most common initial complaints. Other symptoms include nausea, vomiting, malaise, drowsiness, and inability to concentrate. Increasing exposures to CO can cause confusion, shortness of breath, tachycardia, chest pain, loss of consciousness, seizures, and death.1
Pathophysiology
Carbon monoxide is a colorless, odorless gas that is produced during the combustion of fossil fuels. It is the most common cause of poisoning-related deaths (1,000 to 2,000 deaths per year) in the United States.1 The most common sources of CO exposure are furnaces, automobile exhaust, charcoal fires, and gas-powered tools.1
CO binds to hemoglobin with a much higher affinity than oxygen, thus reducing the oxygen-carrying capacity of the blood. CO also shifts the oxyhemoglobin saturation curve leftward, resulting in a further decrease in oxygen delivery to tissues. CO acts by a number of other mechanisms, including binding to cytochrome aa3 with resultant impairment of production of adenosine triphosphate (ATP), generation of oxygen free radicals, lipid peroxidation in the brain, and inducement of cell apoptosis.1 An uncommon source for CO is the chemical methylene chloride, which is metabolized endogenously in humans to form CO.
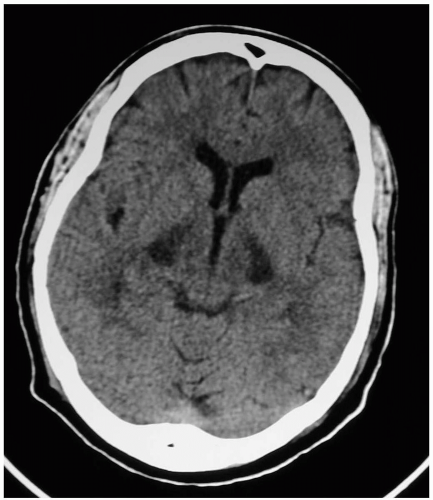 FIGURE 24-10 Bibasilar hypodensities after carbon monoxide toxicity. (Courtesy of Robert Hendrickson, MD.) |
Diagnosis
The diagnosis may be made in most cases by history alone. A carboxyhemoglobin level can be measured by co-oximetry using a venous or arterial blood sample.
Clinical Complications
Patients may develop persistent and delayed neurologic sequelae after CO poisoning. In 25% and 50% of patients who either lose consciousness during exposure to CO or have a carboxyhemoglobin level greater than 25%, symptoms persist for at least 1 month.2 Neurologic problems including ataxia, focal abnormalities, and parkinsonism may occur. Affective symptoms may include depression, sleep disturbance, personality changes, and anxiety. Neuroimaging may show abnormalities in the white matter, hippocampus, and globus pallidus.1
Management
Any patient with suspected toxicity should be immediately placed on high-flow supplemental oxygen to hasten CO elimination. The use of hyperbaric oxygen (HBO) to treat CO poisoning remains controversial, although recent studies provide evidence in favor of this therapy to reduce the incidence of neurologic sequelae.2 HBO significantly increases the elimination of CO, while potentially decreasing CO-induced lipid peroxidation.2 Suggested indications for HBO have included loss of consciousness, persistent neurologic deficits, pregnancy, and carboxyhemoglobin levels greater than 25%.
REFERENCES
1. Weaver LK. Carbon monoxide poisoning. Crit Care Clin 1999;15:297-317.
2. Weaver LK, Hopkins RO, Chan K, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med 2002;347:1057-1067.
24-11 Iron
Anthony Morocco
Clinical Presentation
Iron poisoning may be considered in five stages. Initially, patients develop nausea, vomiting, diarrhea, and abdominal pain and may develop gastrointestinal (GI) hemorrhage and fluid losses. The second stage is a latent phase with resolving GI symptoms that may occur 6 and 24 hours after ingestion. Severe poisonings rapidly progress to a third stage of systemic toxicity, with shock and metabolic acidosis. Stage four occurs 2 to 3 days after ingestion, when hepatic injury is evident. The final stage may occur 2 to 8 weeks after ingestion, with scarring and strictures causing gastric outlet obstruction.1
Pathophysiology
Available iron formulations include ferrous sulfate (28% elemental iron by weight), ferrous fumarate (33% elemental iron), ferrous gluconate (12% elemental iron), ferrous chloride (28% elemental iron), and ferrous lactate (19% elemental iron). Ingestions of greater than 20 mg/kg are considered to be clinically important and potentially harmful.1
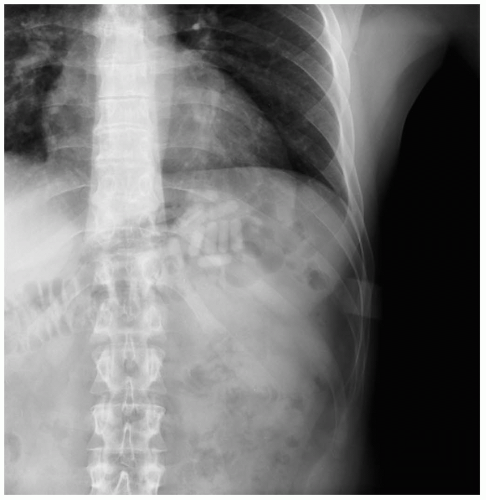 FIGURE 24-11 Multiple iron-containing tablets are seen in the left upper quadrant. (Courtesy of Mark Silverberg, MD.) |
Iron exerts a direct corrosive effect on GI mucosa. Formation of reactive oxygen species causes hepatic injury. Acidosis and cell injury occur by several mechanisms. Conversion of ferrous to ferric iron after absorption to release a hydrogen ion, disruption of oxidative phosphorylation, and direct myocardial toxicity and fluid losses cause hypotension, tissue hypoperfusion, and lactic acidosis.1,2
Diagnosis
Absence of symptoms within 6 hours after ingestion rules out significant toxicity. Iron-containing pills are radiopaque on abdominal radiographs, but chewable and liquid forms are rarely visible. Peak serum iron concentrations occur 2 to 6 hours after ingestion; concentrations greater than 300 µg/dL are associated with GI toxicity, greater than 500 µg/dL with systemic toxicity, and greater than 1,000 µg/dL with severe poisoning and death.1,2,3
Clinical Complications
GI injury may result in bowel edema, ulceration, necrosis, and perforation. Worsening systemic toxicity may result in seizures and coma. Hepatic injury may progress to fulminant failure. Prolonged administration of deferoxamine is associated with acute lung injury and Yersinia enterocolitica sepsis.1
Management
The emergency physician should consider gastric emptying within the first hour after ingestion and whole-bowel irrigation. Activated charcoal does not adsorb well to iron. Aggressive replacement of GI fluid losses is vital. Treatment with the iron chelator, deferoxamine, should be considered for patients with lethargy, intractable vomiting, shock, metabolic acidosis, or a serum iron concentration greater than 500 µg/dL.1,2,3
REFERENCES
1. Fine JS. Iron poisoning. Curr Probl Pediatr 2000;30:71-90.
2. Black J, Zenel JA. Child abuse by intentional iron poisoning presenting as shock and persistent acidosis. Pediatrics 2003;111:197-199.
3. Riordan M, Rylance G, Berry K. Poisoning in children 3: common medicines. Arch Dis Child 2002;87:400-402.
24-12 Methanol
Anthony Morocco
Clinical Presentation
Patients initially develop mild central nervous system depression, headache, and confusion. Nausea, vomiting, and abdominal pain may occur. Visual disturbances such as blurred vision, photophobia, and “snow field” vision occur after a latent period of 40 minutes to 72 hours.1
Pathophysiology
Methanol is a toxic alcohol that is widely used as an industrial solvent, fuel, and chemical precursor. It can be found in reformulated gasoline and in many home products such as paint thinners, windshield washer fluids, cleaning solutions, and camp-stove fuel. It is well absorbed after oral, dermal, or inhalational exposure.1
Methanol is converted by alcohol dehydrogenase to formaldehyde, which is quickly converted by formaldehyde dehydrogenase to formic acid. This toxic metabolite results in acidosis and inhibits cytochrome c in mitochondria. The optic disc and nerve are particularly susceptible to the latter effect. Cytochrome inhibition results in increased lactate production and exacerbates the acidosis.1
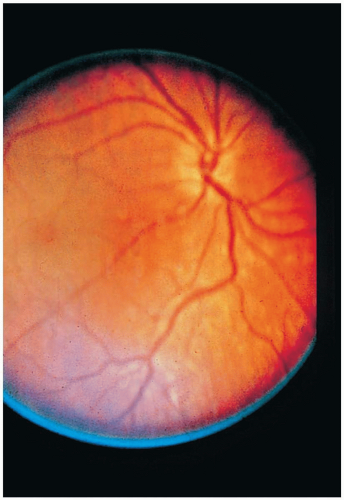 FIGURE 24-12 The right fundus of a commercial sailor who had been at sea for 3 weeks and drank methanol from the ship’s compass. (From Tasman and Jaeger, with permission.) |
Diagnosis
Measurement of serum methanol confirms the diagnosis. Routine laboratory testing reveals an increased osmolal gap and an anion gap metabolic acidosis. Formate levels (not readily available) and low pH correlate well with mortality. Computed tomography and magnetic resonance imaging may show cerebral edema, white-matter changes, and basal ganglia lesions. Eye examination may reveal nystagmus, decreased pupillary response, and hyperemia of the optic disc with subsequent papilledema.1
Clinical Complications
Permanent decreased visual acuity or blindness may occur. Serious poisonings may result in coma and seizures due to cerebral edema and brain injury occurring preferentially in the putamen. Other complications may occur, including pancreatitis, methemoglobinemia, and myoglobinuric renal failure.1
Management
Treatment strategy involves correction of acidosis, inhibition of formate production, and enhancement of methanol and formate clearance. General treatment indications include history of significant ingestion, osmolal gap greater than 10, pH less than 7.3, visual dysfunction, or methanol concentration greater than 20 mg/dL. Bicarbonate administration to maintain a pH greater than 7.3 decreases formate ionization and toxic effects. Ethanol and fomepizole block alcohol dehydrogenase, thus inhibiting formate production. Folinic acid or folic acid administration may enhance formate metabolism. Hemodialysis greatly enhances clearance of formate and methanol and corrects acidosis. Hemodialysis is generally recommended for methanol levels greater than 50 mg/dL, severe acidosis, visual disturbance, or other signs of severe poisoning.1
REFERENCES
1. Barceloux DG, Bond GR, Krenzelok EP, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol 2002;40:415-446.
24-13 Ethylene Glycol
Anthony Morocco
Clinical Presentation
Patients may initially develop symptoms of inebriation, vomiting, and central nervous system depression, or they may appear relatively asymptomatic. Renal failure, acidosis, hypotension, and cerebral edema may occur and may be delayed up to 12 hours or longer.1
Pathophysiology
Ethylene glycol is a toxic alcohol derivative that is widely used in solvents, chemical manufacturing, and antifreeze solutions. It is sweet-tasting and well-absorbed orally, but dermal and inhalational absorptions are poor.1
The first step of ethylene glycol metabolism occurs when ethylene glycol is catalyzed by alcohol dehydrogenase, with further conversion to a number of toxic compounds. Accumulation of glycolic acid results in metabolic acidosis. Direct cellular toxicity and calcium oxalate crystal deposition can cause renal injury.1
 FIGURE 24-13 A: Fluorescence of the urine on the left under a Wood’s lamp. Urine on the right is normal urine. B: Oxalate crystals. (A and B, Courtesy of Robert Hendrickson, MD.) |
Diagnosis
Measurement of serum ethylene glycol confirms the diagnosis. Routine laboratory testing may reveal an increased osmolal gap and an anion gap metabolic acidosis. Hypocalcemia and leukocytosis may also be present. Urinalysis reveals calcium oxalate crystals in up to 50% of patients at admission. Computed tomography scans may show cerebral edema. Because antifreeze solutions often contain fluorescein, examination of urine with a Wood’s lamp may reveal fluorescence and help to confirm a suspicion of ingestion.1
Clinical Complications
Severe poisoning can cause progressive acidosis, hypotension, congestive heart failure, acute respiratory distress syndrome, cerebral edema, seizures, coma, and death.1 Acute tubular necrosis and oliguric renal failure develop 24 to 72 hours after ingestion and may be permanent. Bone marrow depression and cranial nerve dysfunction rarely occurs.1
Management
Aspiration of gastric contents should be considered if the patient presents less than 1 hour after ingestion. Treatment strategy involves correction of acidosis, inhibition of toxic metabolite production, and enhancement of ethylene glycol and toxic metabolite clearance. Ethanol and fomepizole block alcohol dehydrogenase, thus inhibiting toxic metabolite production. Indications for antidotal treatment include history of ethylene glycol ingestion, serum level greater than 20 mg/dL, osmolal gap greater than 10, pH less than 7.3, and presence of urinary oxalate crystals. Sodium bicarbonate should be administered to maintain a pH greater than 7.3. Pyridoxine, thiamine, and magnesium are cofactors for two detoxification pathways. Hemodialysis greatly enhances clearance of ethylene glycol and toxic metabolites and corrects acidosis. Hemodialysis is generally recommended for ethylene glycol levels greater than 50 mg/dL, severe acidosis, renal failure, or other signs of severe poisoning.1
REFERENCES
1. Barceloux DG, Krenzelok EP, Olson K, Watson W. American Academy of Clinical Toxicology practice guidelines on the treatment of ethylene glycol poisoning. J Toxicol Clin Toxicol 1999;37:537-560.
24-14 Isopropanol
Anthony Morocco
Clinical Presentation
Pathophysiology
Isopropanol (isopropyl alcohol) is a chemical that is found in many industrial and household products such as rubbing alcohol (70% isopropanol).1 Isopropanol is well absorbed after oral, dermal, or inhalational exposure.
 FIGURE 24-14 “Blue heaven” (isopropanol). (Courtesy of Robert Hendrickson, MD.) |
Because the inebriating effect of alcohol increases with the number of carbon atoms in its structure, isopropanol has twice the potency of ethanol. Isopropanol is metabolized to acetone by the enzyme alcohol dehydrogenase in the liver. The acetone also has inebriating effects. Metabolism does not produce toxic acids, as occurs with methanol and ethylene glycol exposure.1
Diagnosis
Measurement of serum isopropanol or acetone concentration confirms the diagnosis. Other laboratory abnormalities include an elevated osmolal gap and positive ketones on urinalysis.3 Elevation of the anion gap and acidosis do not occur. The patient may smell strongly of acetone.
Clinical Complications
Large ingestions can cause hypotension due to vasodilation, respiratory depression, and coma. Vomiting may result in aspiration with resultant pneumonia and acute respiratory distress syndrome.1 Hemorrhage gastritis may occur. Other reported effects include renal tubular acidosis, hemorrhagic tracheobronchitis, rhabdomyolysis, and hemolytic anemia.1
Management
REFERENCES
1. Zaman F, Pervez A, Abreo K. Isopropyl alcohol intoxication: a diagnostic challenge. Am J Kidney Dis 2002;40:E12.
2. Stremski E, Hennes H. Accidental isopropanol ingestion in children. Pediatr Emerg Care 2000;16:238-240.
3. Church AS, Witting MD. Laboratory testing in ethanol, methanol, ethylene glycol, and isopropanol toxicities. J Emerg Med 1997;15:687-692.
24-15 Lead
Anthony Morocco
Clinical Presentation
Acute ingestion of large doses of lead may result in altered mental status reflective of encephalopathy within hours. Chronic lead toxicity may have a more subtle presentation, with anorexia as one of the earliest symptoms. Other symptoms may include nausea, vomiting, intermittent abdominal pain, constipation, irritability, distractibility, impulsiveness, encephalopathy, headache, tremor, fatigue, arthralgias, and anemia.
Pathophysiology
Lead is a heavy metal that commonly causes toxicity, particularly in children. Common sources of lead intoxication include ingestion of residue and chips of lead-based paint, occupational exposure, environmental contamination by leaded gasoline and industrial fallout, use of cookware with glazes containing lead oxide (greta), retained bullets, and folk remedies containing greta and azarcon (lead tetroxide).1
Diagnosis
The blood lead concentration should be measured in all suspected cases. Laboratory studies may reveal a microcytic, hypochromic anemia and basophilic stippling of red blood cells on peripheral blood smear. Long-bone radiographs in children may reveal opacities at metaphyses (“lead lines”) due to abnormal bone growth (not lead deposition). Radiography may reveal unabsorbed lead in the gut.1
Clinical Complications
Acute, large lead ingestions may result in hemolysis and hepatorenal failure. Chronic exposure may result in cognitive deficits in some children and hypertension, peripheral neuropathy (i.e., wrist drop), and renal insufficiency in some adults. Patients with high lead concentrations may develop encephalopathy, seizures, and cerebral edema. Lead is teratogenic and may be associated with preterm delivery, low birth weight, and developmental delay.1
Management
Gastric lavage and whole-bowel irrigation should be considered if lead remains in the gut. Symptomatic patients should receive chelation therapy, but the concentration at which to chelate an asymptomatic child (generally recommended at 45 µg/dL) is controversial. Levels greater than 70 µg/dL or significant symptoms may necessitate hospitalization and aggressive chelation. Oral succimer (DMSA) may be used in patients without serious gastrointestinal toxicity. Ethylenediaminetetraacetic acid (EDTA) and dimercaprol (BAL) are parenteral chelators and may be used in cases of encephalopathy, highly elevated blood lead concentration (greater than 70 µg/dL in children or 100 µg/dL in adults), or severe gastrointestinal symptoms.1 BAL and EDTA can have substantial side effects, including hypertension, renal failure, and anaphylaxis; they should be used with caution.
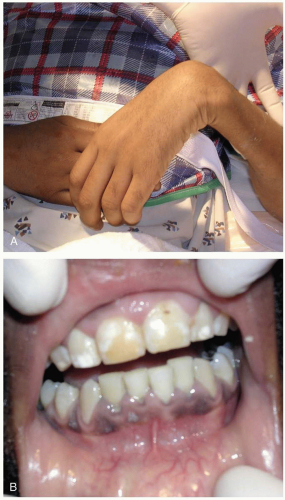 FIGURE 24-15 A: Wrist drop in a patient with elevated blood lead levels. (Courtesy of Rachel Haroz, MD.) B: The gingival discoloration represents “lead lines” in this man who presented with chronic lead intoxication from a bullet retained in his neck due to a gunshot wound 20 years previously. (Courtesy of Judith Eisenberg, MD, Rachel Haroz, MD, and Greg Schneider, MD.) |
REFERENCES
1. Graeme KA, Pollack CV Jr. Heavy metal toxicity, part II: lead and metal fume fever. J Emerg Med 1998;16:171-177.
2. Markowitz M. Lead poisoning. Pediatr Rev 2000;21:327-335.
24-16 Retained Bullets
Anthony Morocco
Clinical Presentation
Patients may present with symptoms of lead toxicity days to years after the initial injury. Complaints may include nausea, constipation, irritability, headache, and insomnia.1
Pathophysiology
Lead bullets and lead-containing fragments often remain in the body after survival from gunshot injuries. In this setting, lead may subsequently be absorbed into the systemic circulation, representing a source for potential toxicity.2
Lead fragments are generally isolated from the surrounding tissue by the formation of a fibrous capsule, and most patients consequently do not develop lead toxicity. However, contact with joint spaces, synovial tissue or membranes, cerebrospinal fluid, or vascularized cystic structures may result in dissolution of lead-containing fragments and substantial lead absorption. The relatively acidic pH, in conjunction with hyaluronic acid in synovial fluid, contributes to this effect. Absorption is more pronounced in joints with increased mobility or weight bearing, and in individuals with chronic inflammatory conditions such as arthritis. Retained shrapnel, fragmented bullets, and buckshot have higher surface areas of exposed lead and therefore may be associated with increased lead absorption. Prolonged time of exposure may also increase lead absorption.1,2
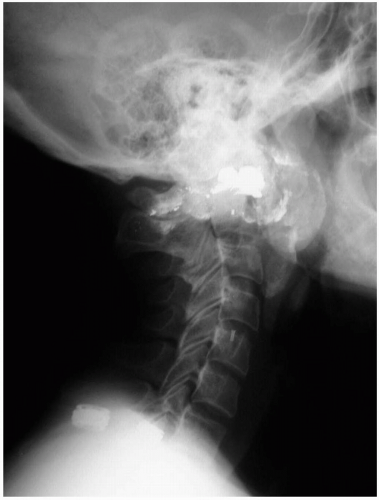 FIGURE 24-16 Retained bullets that are in contact with synovial, peritoneal, or cerebrospinal fluid may produce elevations in serum lead concentration many years after the injury. (Courtesy of Robert Hendrickson, MD.) |
Diagnosis
The potential for lead toxicity can be assessed by measuring the blood lead concentration (BLC). Plain radiography and computed tomographic scanning may help to assess the precise location of lead fragments and aid in removal strategies. Microcytic, hypochromic anemia and basophilic stippling of red blood cells may be evident on peripheral blood smear.1
Clinical Complications
Chronic lead exposure results in lead sequestration in bone. Systemic illness (e.g., sepsis, thyrotoxicosis), surgery, or pregnancy increases bone turnover and releases lead from bony stores.1 Elevated BLCs may be associated with the development of cerebral edema, encephalopathy, and seizures. Chronic lead toxicity may be associated with the development of hypertension, peripheral neuropathy, and renal insufficiency in some patients.1,2
Management
Strong consideration should be given to surgical removal of lead-containing fragments, especially if they are in contact with joint spaces or cerebrospinal fluid. Symptomatic patients may require chelation with parenteral dimercaprol (BAL) and ethylenediaminetetraacetic acid (EDTA), with or without oral succimer (DMSA). If lead fragments are not amenable to surgical removal, management can be problematic, and careful and frequent monitoring of BLCs is required.
REFERENCES
1. McQuirter JL, Rothenberg SJ, Dinkins GA, et al. The effects of retained lead bullets on body lead burden. J Trauma 2001;50:892-899.
2. Farrell SE, Vandevander P, Schoffstall JM, et al. Blood lead levels in emergency department patients with retained lead bullets and shrapnel. Acad Emerg Med 1999;6:208-212.
24-17 Arsenic
Anthony Morocco
Clinical Presentation
Within minutes to hours after acute arsenic ingestion, patients may develop nausea, vomiting, abdominal pain, and profuse watery diarrhea. Cardiovascular effects may include hypotension, endothelial damage, pulmonary edema, prolonged QT interval on electrocardiography, and dysrhythmias. Severe cases may manifest with delirium and seizures.1 Chronic arsenic poisoning may manifest with encephalopathy, peripheral neuropathies, and dermatitis. Chronic skin lesions may include abnormal pigmentation and hyperkeratosis, particularly on the palms and soles. The hallmark of arsine gas exposure is hemolysis, and patients who survive long enough to reach the emergency department may present with various sequelae of hemolysis.1,2
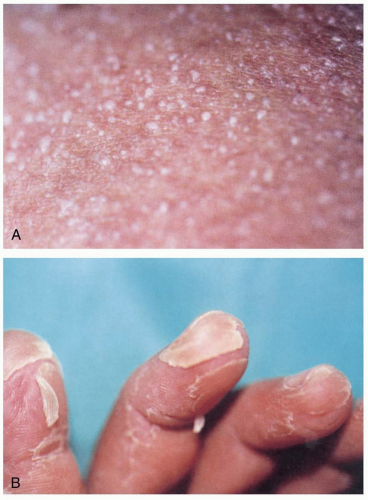 FIGURE 24-17 A: A group of pustules measuring 1 to 3 mm in diameter is seen on erythematous skin on the abdomen. B: Lamellar desquamation and hyperkeratosis are evident on the fingers. (A and B, From Bartolomé B, Córdoba S, Nieto S, et al. Acute arsenic poisoning: clinical and histopathologic features. Br J Dermatol 1999;141:1106-1109, with permission.) |
Pathophysiology
Arsenic exists in several forms: elemental arsenic, organic arsenic, inorganic arsenate (trivalent), inorganic arsenite (pentavalent), and arsine gas. Arsenic is commonly found in the environment (soil and ground water). It is used in some medicines (arsenic trioxide for leukemia), pesticides, chemical warfare agents (lewisite), semiconductor manufacturing (arsine), and wood preservatives and has a myriad of industrial uses.1 Seafood contains predominantly nonharmful organic arsenic compounds.1,2
Arsenic compounds may be absorbed by oral, dermal, or inhalational routes. Arsenic binds to sulfhydryl groups, thereby interfering with a number of enzymes and structural proteins. Arsenate substitutes for phosphate and uncouples oxidative phosphorylation. Arsine binds to red blood cells and causes hemolysis.1,2
Diagnosis
The odor of garlic may be noted on or about the patient. Arsenic in the gastrointestinal tract may be radiopaque. Peripheral blood smears may show basophilic stippling or rouleaux formation.1 Urinary arsenic should be measured, preferably in a 24-hour specimen collected in acid-free containers. Special laboratory techniques can differentiate organic from inorganic forms of arsenic.2
Clinical Complications
Complications of acute arsenic poisoning include seizures, coma, peripheral neuropathy, cardiac arrhythmia, severe fluid loss, shock, liver injury, acute respiratory distress syndrome, pancytopenia, seizures, coma, and death. Complications of chronic arsenic exposure include basal cell and squamous cell carcinoma and lung cancer. Severe anemia and renal failure can complicate arsine gas exposure.1,2
Management
Aggressive supportive care is essential. Whole-bowel irrigation may help clear any arsenic that remains in the gastrointestinal tract. Acute toxicity may be treated with intramuscular dimercaprol (BAL), a chelating agent. Chronic or subacute toxicity may be treated with the oral chelator succimer (DMSA). Chelation usually is not effective for arsine poisoning.1,2
REFERENCES
1. Graeme KA, Pollack CV Jr. Heavy metal toxicity, part 1: arsenic and mercury. J Emerg Med 1998;16:45-56.
2. Ratnaike RN. Acute and chronic arsenic toxicity. Postgrad Med J 2003;79:391-396.
24-18 Mercury
Anthony Morocco
Clinical Presentation
Manifestations of toxicity vary, depending on the type of mercury and the route of exposure. Exposure to elemental mercury may cause fever, cough, shortness of breath, vomiting, diarrhea, and weakness. Ingested mercury salts may cause severe gastrointestinal irritation. Acrodynia (“pink disease”) is a hypersensitivity syndrome of rash and neurologic symptoms that usually is seen in children exposed to mercury salts.1 Elemental and inorganic mercury can cause subacute or chronic mercury toxicity. Patients may be upset at presentation, with loose teeth, renal dysfunction, intention tremor, and personality changes such as shyness (erethism), depression, and fatigue (neurasthenia). The short-chain organic compounds methylmercury and dimethylmercury cause acute dermatitis and gastrointestinal injury, with severe neurologic dysfunction developing over days to weeks.
Pathophysiology
Mercury is a heavy metal that may exist in an elemental, inorganic salt, or organic form.1 Elemental mercury is widely available and may be found in some scientific instruments, thermostats, thermometers, and dental amalgams. Mercury salts may be found in some disc batteries, disinfectants, and various industrial processes. Organic mercury may be encountered in laboratories, pesticides, and wood preservatives.
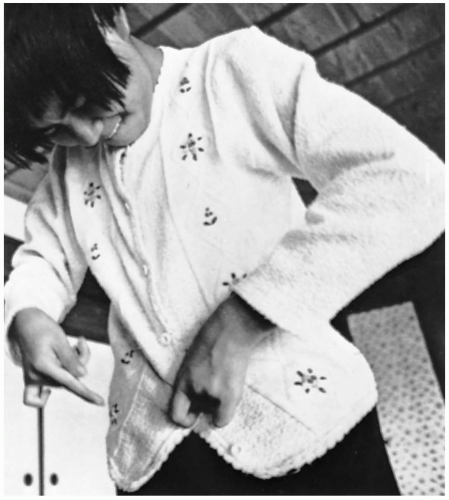 FIGURE 24-18 Minamata victim: severe ataxia of upper limb displayed by buttoning. (From Putnam JJ. Quicksilver and slow death. Natl Geog 1972;142:507, with permission.) |
Elemental mercury is best absorbed by inhalation; absorption in a normal gastrointestinal tract is negligible. Organic mercury and mercury salts may be absorbed orally or dermally. Mercury binds to sulfhydryl, phosphoryl, and other groups, causing disruption of enzymes and structural proteins and resultant tissue damage.1
Diagnosis
The diagnosis may be confirmed by measurement of blood mercury concentration (acute toxicity) or a 24-hour urine mercury concentration (chronic toxicity), or both.1
Clinical Complications
Intravenous administration of mercury reportedly may cause pulmonary emboli, and subcutaneous injection may cause aseptic abscesses and granulomas.2 Acute mercury inhalation can result in respiratory failure and death. Mercuric salt ingestion can cause renal failure. Shock and death may occur from hemorrhagic gastroenteritis and fluid loss. Under certain circumstances, exposure to organic mercury, particularly dimethylmercury, has caused progressive, irreversible, and sometimes fatal central nervous system damage.3
Management
REFERENCES
1. Graeme KA, Pollack CV Jr. Heavy metal toxicity, part 1: arsenic and mercury. J Emerg Med 1998;16:45-56.
2. Ruha AM, Tanen DA, Suchard JR, et al. Combined ingestion and subcutaneous injection of elemental mercury. J Emerg Med 2001;20:39-42.
3. Nierenberg DW, Nordgren RE, Chang MB, et al. Delayed cerebellar disease and death after accidental exposure to dimethylmercury. N Engl J Med 1998;338:1672-1676.
24-19 Thallium
Anthony Morocco
Clinical Presentation
Symptoms develop hours to days after exposure. The classic presentation of thallium toxicity is the combination of alopecia and a painful, ascending peripheral neuropathy. The neuropathy begins in the lower extremities 2 to 5 days after exposure, and patients may experience severe pain with light touch. Central nervous system involvement results in a variety of problems, including ataxia, delirium, psychosis, sleep disturbance, optic neuropathy, and cranial nerve dysfunction. Alopecia begins approximately 10 days after exposure; it may be complete or may spare axillary and facial hair. Other signs and symptoms include nausea, vomiting, constipation, abdominal pain, tachycardia, hypertension, palmar erythema, anhydrosis, and Mee’s lines.1
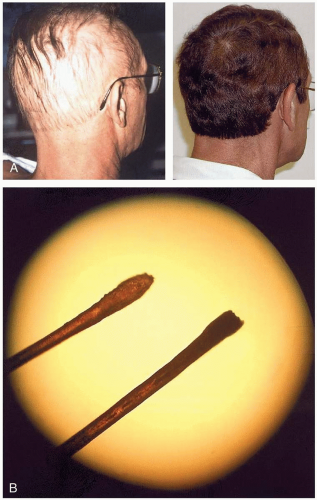 FIGURE 24-19 A: On the left is a patient with thalliuminduced alopecia. On the right is the same patient 6 months later. B: Blackened hair root on the right compared with normal hair on the left. (A and B reprinted from Rusyniak with permission.) |
Pathophysiology
Thallium is a metal that may be found in its elemental form or as a salt. Although elemental thallium is usually harmless, the thallous (Tl+1) and thallic (Tl+3) salts are potentially harmful. Thallium salts such as thallium sulfate have been used in depilatories, antimicrobial agents, and rodenticide agents. The use of thallium salts as rodenticides was banned in the United States in 1975 but persists elsewhere.1
Thallium salts are well absorbed by oral, dermal, and inhalational routes. Thallium inhibits a number of potassium-dependent enzymes and disrupts Krebs cycle enzymes, sodium-potassium adenosine triphosphatase (Na+/K+-ATPase), mitochondrial enzymes, and others. Cells of the central and peripheral nervous system are particularly affected, resulting in numerous lesions, including axonopathy.
Diagnosis
Thallium poisoning should be strongly suspected in any patient with alopecia and peripheral neuropathy. The metal can be seen under a microscope as black deposits in hair roots in 95% of patients, in several bands if multiple exposures have occurred. Abdominal radiography may reveal metallic material in the gastrointestinal tract. Laboratory confirmation of thallium toxicity involves measurement in a 24-hour urine collection.
Clinical Complications
Patients with severe toxicity progressively worsen over days, ultimately to coma, respiratory paralysis, and death.
Management
Multidose activated charcoal decreases thallium absorption and enhances elimination. Whole-bowel irrigation should be considered, especially if thallium remains in the gastrointestinal tract. Prussian blue (ferric hexacyanoferrate) should be administered as soon as possible, because it binds thallium and enhances elimination.1,2 Prussian blue may not be readily available, but substantial supplies are maintained by the Radiation Emergency Assistance Center Training Site (REAC/TS) in Oak Ridge, Tennessee (telephone, 1-865-576-1005). Practitioners may contact REAC/TS for guidance in obtaining this antidote. Charcoal hemoperfusion with or without hemodialysis may be beneficial.
REFERENCES
1. Galvan-Arzate S, Santamaria A. Thallium toxicity. Toxicol Lett 1998;99:1-13.
2. Hoffman RS. Thallium poisoning during pregnancy: a case report and comprehensive literature review. J Toxicol Clin Toxicol 2000;38:767-775.
24-20 Methemoglobinemia
Anthony Morocco
Clinical Presentation
Patients with chronic methemoglobinemia may be asymptomatic despite large fractions of methemoglobin. Patients with acute methemoglobinemia, as occurs after exposure to a medication, may present with signs and symptoms of hypoxemia. Patients may develop dyspnea, tachycardia, cyanosis, and nausea as the methemoglobin fraction rises to 30% or more of total hemoglobin. As the methemoglobin fraction rises to greater than 50%, symptoms worsen, mental status may become altered, and hypotension may develop.1
Pathophysiology
Normal hemoglobin is converted to methemoglobin when its iron atom is oxidized from the ferrous (Fe2+) to the ferric (Fe3+) state, which renders it unable to bind oxygen. Chronic methemoglobinemia results from an enzyme deficiency and inherited abnormal forms of hemoglobin, although a small amount of methemoglobin is present in all individuals. Large concentrations of methemoglobin may be produced on exposure to oxidizing agents such as nitrates, nitrites, local anesthetics, chlorates, naphthalene, phenazopyridine, and dapsone.1 Methemoglobinemia may also occur in young infants with diarrhea, dehydration, and acidosis.1
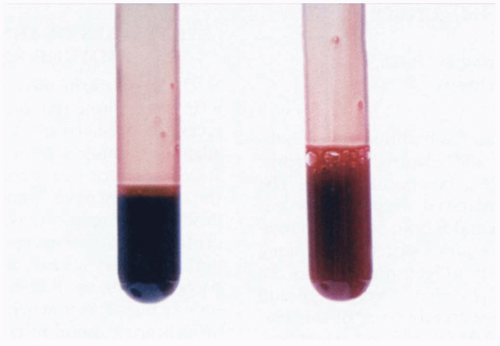 FIGURE 24-20 Chocolate-brown blood on left (methemoglobinemia) and normal blood on right. (From Donnelly R. Methemoglobinemia. N Engl J Med 2000;343:337. © 2000 Massachusetts Medical Society. All rights reserved.) |
Diagnosis
The blood from affected patients appears dark brown in color. The methemoglobin concentration can be measured directly by a co-oximeter. Pulse oximetry is unreliable when methemoglobin is present and reveals a “saturation gap.” The partial pressure of oxygen (pO2) on an arterial blood gas measurement is elevated, and therefore the saturation, which is calculated from the pO2, is falsely elevated. The bedside pulse oximeter may overestimate the arterial hemoglobin oxygen saturation because it falsely reads methemoglobin as oxygenated hemoglobin. For this reason, patients with methemoglobinemia rarely have bedside pulse oximeter readings lower than 85%.1
Clinical Complications
A methemoglobin fraction greater than 70% is reported to be lethal. Other complications include cardiac dysrhythmias, hypotension, hemolysis, and renal failure.1
Management
Treatment is indicated for symptomatic patients with greater than 20% methemoglobin and for asymptomatic patients with greater than 30% methemoglobin.1 Methylene blue, 1 to 2 mg/kg, is given by slow intravenous infusion. This agent accelerates the natural conversion of methemoglobin to hemoglobin. Methylene blue is contraindicated in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Multiple courses of treatment may be needed, particularly after overdose from drugs with a long half-life, such as dapsone.1
REFERENCES
1. Rehman HU. Methemoglobinemia. West J Med 2001;175:193-196.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


