KEY POINTS
Virtually all patients admitted to an ICU have low levels of serum triiodothyronine (T3), and 30% to 50% have low levels of thyroxine (T4) with normal or low levels of serum thyrotropin (TSH).
Patients who have a T4 level of less than 3.0µg/dL despite normal levels of T4-binding proteins have a 68% to 84% mortality rate.
T3 is the logical choice for critically ill patients requiring thyroid hormone replacement.
Early intubation and mechanical ventilation are crucial for successful treatment of myxedema coma.
Management of myxedema coma should include administration of glucocorticoids while the adrenal status is being assessed.
Alterations in thyroid function change the metabolism of almost all drugs, and the doses need careful adjustment to prevent drug toxicity or decreased efficacy.
Autonomous hypersecretion and exogenous overdose of thyroid hormone are the most common causes of severe thyrotoxicosis.
Hyperpyrexia and altered mental status are the hallmarks of thyroid storm.
Medical treatment of severe hyperthyroidism usually normalizes circulating thyroid hormone levels in 2 to 3 weeks, except under circumstances of iodine overload, in which case hyperthyroxinemia may persist for months.
Blockade of hormonal secretion is best accomplished by the addition of stable iodine to an antithyroid drug regimen.
In severe thyrotoxicosis, treatment with iopanoic acid can be lifesaving.
β-Blockers prevent thyroid storm in the thyrotoxic patient undergoing surgery, and they may ameliorate cardiovascular dysfunction in thyroid storm, but their side effects often interfere with therapy in the elderly, in patients with asthma, and in patients with cardiomyopathy.
Amiodarone-induced thyrotoxicosis in a critically ill patient should be managed with methimazole (30-50 mg/d), potassium perchlorate (500 mg twice a day), and prednisone (30-40 mg/d).
After gastric aspiration and lavage, only symptomatic and supportive treatment is needed in cases of levothyroxine overdose.
Neonatal thyrotoxicosis can be life threatening; it is usually caused by transplacental transfer of thyroid-stimulating antibodies. It is transient and requires only short-term treatment.
HYPOTHYROIDISM, NONTHYROIDAL ILLNESS, AND MYXEDEMA COMA
Hypothyroidism is a state of tissue deprivation of thyroid hormone. It is manifested by general reduction of the metabolic rate accompanied by specific symptoms and signs. Usually, hypothyroidism is caused by a decreased supply of thyroid hormone due to one of the following: (1) failure of the gland to synthesize and secrete thyroid hormone, (2) failure of the pituitary to secrete thyrotropin (thyroid-stimulating hormone [TSH]), or (3) hypothalamic disease resulting in a deficiency of thyrotropin-releasing hormone (TRH).
Perhaps the most controversial, if not the most challenging, aspects of thyroidology for the intensivist are how to interpret thyroid function tests in critically ill patients and what to do when the test results are abnormal. Clinically important hypothyroidism in its most severe form usually is seen in patients with primary hypothyroidism who then develop some intercurrent illness; it develops over several weeks and culminates in myxedema coma. Equally challenging are the thyroid function abnormalities seen in patients with concurrent severe illness and the assessment of the thyroid hormone status at the tissue level.
EVALUATION OF THYROID FUNCTION IN PATIENTS WITH SEVERE NONTHYROIDAL ILLNESS SYNDROME (NTIS)
Virtually all critically ill patients have reduced serum levels of triiodothyronine (T3), and approximately 30% to 50% also have low levels of thyroxine (T4), both associated with normal or low serum TSH values.1 This phenomenon has been termed low T3 syndrome, nonthyroidal illness (NTI), or euthyroid sick syndrome. Each of these descriptive terms assumes a priori that such patients are euthyroid despite reduced thyroid hormone levels. The condition is not limited to acute illness. Patients with chronic hepatic or renal failure, calorie deprivation, and a variety of other illnesses present similar thyroid hormone profiles.2 Serum concentrations of both T3 and T4 are also decreased following nonthyroid surgical procedures.3 In patients with a T4 value of less than 3.0µg/dL, the mortality rate is 68% to 84%,4 indicating that a low T4 concentration without a corresponding increase in TSH is a marker for a high risk of death in the critically ill population. Critical illness results in a profound change in set point of the hypothalamic-pituitary-thyroid axis and perturbs local levels of thyroid hormone available to various tissues due to differential metabolism.5 To understand this phenomenon and develop a rational basis for treatment, it is useful to review the thyroid physiology, with emphasis on processes occurring in NTI.
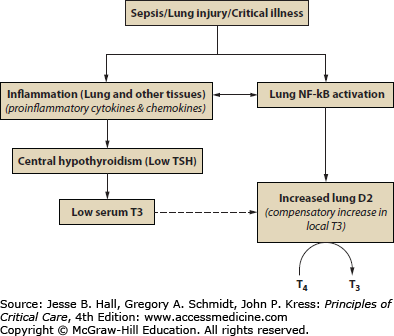
FIGURE 103-1
Physiologic basis for nonthyroidal illness syndrome (NTIS). Diagrammatic representation of the events that may contribute to NTIS, including the effects on the hypothalamus and pituitary and site of thyroid hormone action at the peripheral tissue level. D2, deiodinase type 2; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; T3, triiodothyronine; T4, thyroxine; TSH, thyroid-stimulating hormone. (Barca-Mayo O, Liao XH, DiCosmo C, et al. Role of type 2 deiodinase in response to acute lung injury (ALI) in mice, Proc Natl Acad Sci USA. 2011 Dec 6;108(49):E1321-E1329.)
Metabolism of Thyroid Hormone in Peripheral Tissues: Ninety percent of the hormone secreted by the thyroid gland is T4, the remainder being T3. Thyroid hormone is metabolized in peripheral tissues by stepwise monodeiodination until the molecule is completely stripped of iodine. This process uses specific enzymes called deiodinases.91 The deiodination of T4 can take one of two pathways—removal of the iodine from the outer or phenolic ring (5 position), resulting in 3,3′, 5-triiodothyronine (T3) or removal of the iodine from the inner or tyrosyl ring (5 position), yielding 3,3′, 5′-triiodothyronine (reverse T3 [rT3]). T3 is the active form of the hormone, whereas rT3 has no biologic activity. The same enzyme that removes iodine from the 5′ position of the T4 molecule is also responsible for deiodination of the 5′ iodine from rT3. Therefore, reduction in the 5′-deiodinase activity, which is invariably associated with severe illness and malnutrition, not only reduces the serum T3 level but also increases that of rT3.
The deiodinases are selenium-containing enzymes and dependent on selenium to function. In critical illness, it has been reported that selenium levels have a negative correlation with the APACHE II scores and patients with a score >15 had a marked decrease in selenium levels.6 Several pharmaconutrition studies have suggested that selenium supplementation may improve outcomes from critical illness.7
Thyroid Physiology in the Brain in NTIS: The nature of perturbations of the hypothalamic-pituitary-thyroid axis in critically ill patients is beginning to be understood. The basal TSH values in serum of humans with NTIS can be normal or low, but the response of TSH to TRH usually is attenuated.2 Stress and malnutrition may be partly responsible. In rats, starvation reduces hypothalamic messenger ribonucleic acid (mRNA) for TRH, reduces portal serum TRH, and lowers pituitary TSH content.8 Furthermore, low TRH mRNA has been documented in the paraventricular nuclei of patients with NTIS.9 One therefore would predict that if diminished TRH is, at least in part, contributing to NTIS and the low thyroid hormone levels, then treatment with TRH should increase the TSH and therefore increase the thyroid hormone levels. In fact, Van den Berghe and colleagues10 have demonstrated that administration of TRH to patients with NTIS leads to increase in serum TSH, T3, and T4 levels. Critically ill patients who eventually will recover from their illness have, as a rule, less impairment of the TSH response to TRH. The mechanism for the reduced TRH production in NTIS is unknown but may be related to cytokines or glucocorticoids. These factors probably are responsible for suppression not only of TRH but also of other hypothalamic factors that may be decreased in NTIS, such as corticotropin-releasing hormone and gonadotropin-releasing hormone.
The preceding, however, does not rule out an effect of NTIS directly on the pituitary. For example, why is serum TSH not elevated when the thyroid hormone levels are low? There is experimental evidence that the pituitary may be “euthyroid” owing to increased pituitary conversion of T4 to T3, unlike other tissues.11
In addition, drugs commonly administered to ICU patients have inhibitory effects on the hypothalamic and pituitary function. Dopamine is one such drug; it inhibits TSH even when infused at low doses.12,13
Other Actions of Thyroid Hormone on Physiology in NTIS: Type II pneumocytes, which have been shown to be involved in regulation of lung function, express thyroid hormone receptors on their surfaces.14 Furthermore, T3 has been shown to modulate surfactant function during sepsis.15-17
Sepsis and multisystem organ failure often are associated with disseminated intravascular coagulation (DIC) and consumption of coagulation inhibitors such as antithrombin III. Rats with experimentally induced NTIS treated with T3 show attenuation of sepsis-induced decreases in antithrombin III levels.18
The levels of thyroid hormone and TSH are measured in many critically ill patients at some point during the course of hospitalization. Low concentrations of thyroid hormone without an appropriate increase in serum TSH level would, under normal conditions, raise the suspicion of pituitary (secondary) or hypothalamic (tertiary) hypothyroidism. However, in a critically ill patient, the diagnosis of primary hypothyroidism with inadequate pituitary response needs consideration. A modest elevation of serum TSH level without an increase in rT3 concentration is a strong indicator of primary hypothyroidism. Except in the presence of renal failure, a decreased rT3 level raises the possibility of hypothyroidism and should prompt a search for the etiology. While these possible diagnoses are being investigated, thyroid hormone replacement and glucocorticoid treatment are indicated. Since results of rT3 measurement often are not obtained readily, this test is useful in retrospect for ruling out primary endocrine dysfunction and adds little to the initial management of patients. Since only severe primary hypothyroidism requires emergency treatment, its recognition is usually simple because of persistent TSH elevation, a prior history of thyroid disease, and physical findings compatible with hypothyroidism.
Should patients with low serum levels of thyroid hormone in the face of catastrophic NTIS receive hormonal replacement? In order to answer this question, several issues need to be determined: (1) Is the serum TSH level an accurate reflection of the thyroid status of all body tissues, or does it only reflect the status of the pituitary? (2) Are the tissues of the body functionally hypothyroid? and (3) Is the low serum T3 level an adaptive mechanism for conservation of energy during critical illness? Unfortunately, these questions remain relatively unanswered. In a randomized, prospective study to determine the effect of T4 treatment in NTIS, 11 patients admitted to an ICU with reduced thyroid hormone levels were treated with intravenous T4, and 12 served as controls.19 The study indicated an earlier mortality in the treated group, although the number of survivors was not significantly different between the groups. It was concluded that T4 therapy was not beneficial, and inhibition of TSH secretion by the administration of T4 may be detrimental to the recovery of thyroid function. These results have been confirmed in a similar study carried out in an ICU setting.20 Nevertheless, many physicians understandably find it difficult to withhold treatment in a dying patient with virtually undetectable thyroid hormone levels.
Not only are data on the benefit of thyroid hormone treatment limited, but it is unlikely that an answer regarding the efficacy of this treatment will be forthcoming, given multiple-organ involvement in serious illness and the difficulty of interpreting the effect of thyroid hormone replacement in individuals receiving multiple drugs. Thus the argument centers not only on the question of whether such patients are truly hypothyroid (hence the bias of the term euthyroid sick) but also on whether this temporary hypothyroidism may not, in fact, be beneficial. Inhibition of the type I 5′-deiodinase is the principal mechanism that reduces the supply of biologically active thyroid hormone, T3, to peripheral tissues in the severely ill. Experimental work in a rat model indicates that peripheral tissue hypothyroidism is maintained by an inhibition of the usual compensatory increase in TSH level. This inhibition occurs because normal levels of T3 are generated in the pituitary gland, which uses a different form of 5′-deiodinase, the type II enzyme, that is actually more active in severe illness.11,21 Teleologists argue that this mechanism to reduce the delivery of thyroid hormone to peripheral tissues is not accidental and, therefore, that reduced metabolic activity may be beneficial in the face of the increased catabolism characteristic of severe illness. The question is: Does the physician or nature know best? At our institution there is no consensus on this subject.
Studies have shown decreased serum T3 concentrations in patients undergoing cardiopulmonary bypass surgery. T3 treatment can normalize the serum levels and may increase cardiac output and lower systemic vascular resistance22; however, these effects may be negligible.23
Table 103-2 may serve as a bedside guide to the intensivist for the selection of patients for thyroid hormone treatment.
Indications for Thyroid Hormone Treatment in Patients with Severe NTI
| Increased serum TSH concentrations |
| History of radioactive iodine treatment |
| Hypothermia |
| Macroglossia |
| Goiter |
| History of thyroid disease |
| Treatment with thyroid hormone at any time prior to the current illness |
| Hypercholesterolemia |
| Hyporeflexia |
| Unexplained pleural or pericardial effusions |
| Increased serum creatine phosphokinase level |
If the decision is made to treat a sick patient who has reduced thyroid hormone levels, the logical choice is T3. Administration of T4 does not change the serum T3 concentration significantly—it only increases the level of the biologically inactive rT3. The problem with T3 treatment is its potential cardiac “toxicity.” One possible problem is the proarrhythmogenic effect of T3 in anesthetized patients; another possible problem is that T3 may increase the oxygen demand of a myocardium with a fixed coronary artery lesion. T3 is now available as the product Triostat™; however, the cost of a single day’s treatment for myxedema would be in excess of $3500! Alternatively, a solution of T3 for intravenous use can be prepared by the hospital pharmacist by dissolving l-T3 in 0.1 N NaOH, followed by a 10-fold dilution in normal saline containing 2% albumin to a final concentration of 25µg T3 per milliliter. The solution is sterilized by a single passage through a 0.22-µm Millipore filter and is stored, for no longer than 1 week, at 4°C, protected from light. The materials for this preparation cost about $1.
MYXEDEMA COMA
Myxedema coma is caused by marked and prolonged depletion of thyroid hormone. The cardinal features of myxedema coma are (1) defective thermoregulation to the point of hypothermia, (2) altered mental status to the point of coma, (3) a history or sign (such as a neck scar) of ablative thyroid treatment, and (4) an identifiable precipitating event. The condition is a medical emergency because it is fatal in approximately one-half of cases.24 This rare entity occurs typically in elderly women with long-standing hypothyroidism who develop an intercurrent illness and lapse into coma or in hypothyroid persons exposed to cold or given sedatives, hypnotics or opiates, sufficient to push them to the brink of myxedema coma. Table 103-3 lists events likely to precipitate myxedema coma. The usual features of severe hypothyroidism (myxedema) include dry, coarse skin, scaly elbows and knees, yellowness in the skin without scleral icterus, coarse hair, thinning of the lateral aspect of eyebrows, macroglossia and hoarseness, obtundation, delayed deep tendon reflexes, and hypothermia. When a markedly reduced serum T4 level and an elevated TSH concentration accompany these signs, the diagnosis is obvious. However, as for thyrotoxic crisis, initiation of treatment should not be delayed until the results of the thyroid function studies are available. Furthermore, because of intercurrent illness, TSH values may not be elevated in proportion to the severity of hypothyroidism. A high index of suspicion in a patient presenting as just described should prompt immediate treatment after a blood sample is taken for laboratory confirmation of the diagnosis.
Alveolar hypoventilation is known to occur in myxedema.25 It is thus not surprising that patients with underlying lung pathology experience worsening of gas exchange. It has been demonstrated that the hypoxic ventilatory drive is depressed in patients with myxedema and that it responds to hormone replacement.26 The hypercapnic ventilatory response is also significantly depressed, but it does not change with replacement of thyroid hormone. Therefore, a reduced central nervous system (CNS) drive to breathe and decreased respiratory muscle activity are the main reasons for respiratory depression in myxedema coma. Secondary aspiration pneumonia, laryngeal obstruction, and reduced surfactant contribute to lung dysfunction. It is important to be alert to the potential for subtle but progressive aspiration and ventilatory failure.
The cardiovascular complications in myxedema coma are caused by the combination of hypothyroid cardiomyopathy, hypothermia, and hypoxia. Pericardial effusion is almost a constant finding, but it rarely leads to tamponade. It is best demonstrated by echocardiography. In patients with long-standing hypothyroidism, hypercholesterolemia may accelerate the progress of atherosclerosis, leading to ischemic heart disease. The reader is referred to the discussion of hypothermia and its cardiovascular complications (see Chap. 131).
The intercurrent illness and decreased food intake caused by the mental obtundation of myxedema may reduce the serum levels of cholesterol and TSH, diminishing their value as indicators of the severity of the myxedema. Patients presenting with a more profound hypothermia have a poor prognosis. The laboratory findings in patients with myxedema coma are listed in Table 103-4.
Thyroid Hormone: Although severe hypothyroidism, especially in elderly patients, should be treated cautiously, with gradual increments of small doses of thyroid hormone, myxedema coma is an exception to this rule. The immediate threat to life takes precedence over the risks of rapid hormone replacement. The advantage of treating critically ill patients with T3 as opposed to T4 was discussed earlier. In hypothyroid patients without major intercurrent illness, T4 therapy alone may be sufficient to increase the serum T3 level to normal in 2 to 3 days. This is unlikely to be true in ICU patients with multiple-organ-system failure. The principle of hormonal treatment is to rapidly replenish the extrathyroid pool of thyroid hormone, which consists mainly of hormone bound to serum proteins, and to provide the tissues with their daily requirement of the biologically active hormone. Replenishment is best achieved by the immediate administration of T4, a hormone with a considerably longer half-life (7 days) and higher affinity for serum proteins than T3.25-27 The active form of the hormone, T3, then can be provided because it is readily available to tissues and carries a smaller risk of accumulating to excessive amounts (owing to a half-life of approximately 1 day).
The average size of the extrathyroid T4 pool is approximately 800µg/1.73 m2.27-30 On the basis of this estimate and the normal turnover rate of 10% per day, the daily T4 requirement can be calculated to be, on average, 80µg (possibly 50µg in hypothyroidism, owing to a reduced rate of hormone degradation). Intensivists using only T4 for treatment should give initially 500µg l-T4, followed by 50 to 100µg daily. The serum T4 concentration should be in the normal range within 24 to 48 hours. Daily electrocardiographic (ECG) monitoring for ischemic changes and continuous monitoring of rhythm are essential.
We prefer a regimen that uses both T4 and T3. Following the intravenous loading dose of 500µg T4, 25µg T3 is given every 6 hours through a nasogastric tube until improvement is noted, and provided the diagnosis has been confirmed by laboratory tests. The dose is then reduced to maintenance level, and the agent is changed to T4 only after recovery from intercurrent illness.
Use of Steroids: The rate of metabolism of most drugs and natural compounds is markedly reduced in patients with myxedema coma. Therefore, the absolute requirement for steroids is reduced. However, because of the 5% to 10% incidence of associated primary hypoadrenalism, glucocorticoids should be given until evidence for intact adrenal function is secured by the cortisol measurement on the blood sample obtained on admission. The usual dose of hydrocortisone is 50 mg intravenously every 6 hours. The steroid dose then can be tapered rapidly after confirmation of a normal pituitary-adrenal axis. Alternatively, the initial dose can be 2 mg dexamethasone, and a 1-hour adrenocorticotropin hormone (ACTH, cosyntropin) stimulation test can be done on the spot to assess adrenocortical function29 (see Chap. 102).
Supportive Care: Early intubation and mechanical ventilation are believed to be central for the successful treatment of myxedema coma. Severe hemodynamic collapse in the presence of a large pericardial effusion may necessitate immediate pericardiocentesis. Because hypothyroidism can cause an elevation of the serum creatine phosphokinase (CPK) level, obtaining a baseline value is helpful for follow-up, particularly if a myocardial infarction is later suspected. Moderate elevations of the blood urea nitrogen (BUN) and creatinine levels are not uncommon and are not necessarily indicative of chronic renal failure.
Hypothermia is treated with blankets, letting internal heat generation slowly warm the body.24 External warming runs the risk of initiating shock by producing peripheral vasodilation in a patient with already reduced cardiac output. Patients with myxedema are rarely volume overloaded, and the use of diuretics runs the risk of further reducing cardiac output. Hyponatremia is best treated by water restriction because the total body sodium content is increased owing to the storage of sodium in glycosaminoglycan, forming the myxedematous accumulation that becomes mobilized with thyroid hormone treatment. Antiulcer prophylaxis is recommended. More important, it should be remembered that hypothyroidism reduces the metabolism of all drugs, and their dosing needs careful adjustment to prevent drug toxicity. Diligent investigation into the precipitating causes should include blood, urine, and sputum cultures, and empirical treatment with antibiotics should be given.
If severe anemia is present, it should be corrected with blood transfusion to increase the oxygen-carrying capacity of the blood. Use of α-adrenergic agents should be avoided because patients are already vasoconstricted.
Assuming that an accurate diagnosis has been made and that proper therapeutic measures have been carried out, how are the patient’s progress and the efficacy of treatment followed? The physician is committed to treat the patient for several days or as long as the serum TSH concentration remains elevated. Reduction of the TSH level is the earliest indicator of a response to thyroid hormone therapy. Irreversible damage to the respiratory centers has been observed, with failure of spontaneous respiration, despite full repletion of thyroid hormone. No laboratory measurements are helpful in assessing the peripheral tissue responses to thyroid hormone in the critical care setting. The ultimate gauge of successful treatment is complete clinical recovery.
GOITER AND ACUTE AIRWAY OBSTRUCTION
Large goiters, weighing 150 g or more, can cause some degree of tracheal obstruction, especially if they are substernal in location. In a series of 2908 goiters, only 58 (2.0%) caused tracheal obstruction at presentation. Tracheal compression obstructing up to 75% of the tracheal lumen often remains asymptomatic.31 Although dyspnea on exertion has been attributed to goiter, the symptoms are often only nocturnal, manifesting as stridor or, when more severe, as sleep apnea. This problem can be confirmed by x-ray and CT views of the trachea at rest and during a reverse Valsalva maneuver and by sleep studies.32
Growth of the goiter would have to be extensive to cause direct tracheal compression. In Riedel struma, there is tracheal cartilage destruction by fibrous invasion, which can also cause bilateral vocal cord paralysis. Several case reports have been published describing the acute presentation of tracheal obstruction associated with goiter.33,34 Management of these patients is somewhat difficult because emergency tracheostomy may be difficult to perform owing to interference by the thyroid gland. The use of small endotracheal tubes and immediate subtotal thyroidectomy should reduce the need for tracheostomy. It should be noted that subtotal thyroidectomy may not be successful in the presence of tracheomalacia, which may necessitate prosthetic supports.35 In some instances, a simple division of the thyroid isthmus may be sufficient to relieve the symptoms. Although not particularly useful in the acute care setting, 131I therapy can be effective in the long term in elderly patients with large, compressive goiters.36








