sustained injury and developed similar skin lesions, although he was only mildly affected and was not brought to Israel. The other children had escaped injury and were healthy. On arrival Hala was a sick child with high fever and extensive skin lesions on her upper and lower legs shown in the illustrations. The lesions were extremely painful and were characterised by sero-mucoid non-purulent discharge. The margins were slightly raised and sometimes had a violaceous hue. She had anaemia and persistent pronounced leucocytosis with neutrophilia, usually in the range of WBC 48–61 × 103/μL (absolute neutrophils 39–50 × 103/μL).
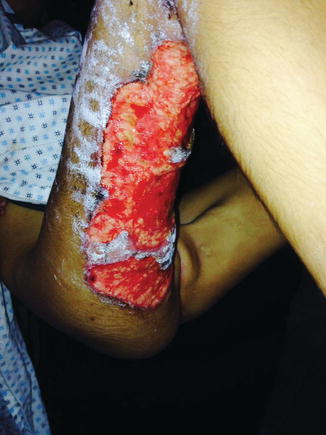
Cultures of the wounds revealed Klebsiella sp. and Escherichia sp., and initial treatment was begun with antibiotics, topical cream and dressing combined with pain relief and nutritional support.
The combined efforts of many skilled professionals were required to save Hala’s life, doctors, surgeons, nurses, nutritionists, physiotherapists, teachers, medical clowns and social workers. Following a period of stabilisation, it was elected to perform skin transplantation in view of the large size of the skin defects. Attempts to do this were tried on two occasions, and unfortunately both attempts failed to generate significant skin healing. It became apparent that the child’s condition was not typical of thermal or chemical burns and that another explanation was likely. Marginal skin biopsies revealed deep lymphohistiocytic and eosinophilic infiltrates together with plasma cells. There was evidence of small vessel vasculitis with necrosis and thromboembolism. Some evidence of patchy fat necrosis was seen. Gram and PAS staining for micro-organisms was negative in some sections, whereas gram-negative bacteria in micro-abscesses were seen in other fields. While not being pathognomonic, this histological picture taken together with the clinical and laboratory features is compatible with a diagnosis of pyoderma gangrenosum with chronic inflammation. Lesions of this kind are unusual in children and maybe related to inflammatory bowel disease, systemic vasculitis, neoplasia, chronic infection as well as toxic, radiation and drug-induced aetiologies. However, history together with extensive investigations which were carried out permitted exclusion of all these causes.
At this time the possibility of an alternative diagnosis was considered. Based on the persistent neutrophilia of the patient, the lack of purulent infection, failure to identify a more common underlying disorder, related parents and possible family history of a similar condition in a brother, a rare genetic cause of pyoderma gangrenosum which could explain the clinical and histological picture was considered. Four reports of children with pyoderma gangrenosum and leukocyte adhesion deficiency type I (LAD I) had been reported in the literature [1–4] by 2013, and subsequently a fifth report has been published [5]. It has never been reported before in patients from the Middle East including Israel.
LAD I is a rare Mendelian autosomal recessive trait caused by mutations in the IGBT2 gene, chromosome 21q22.3 (OMIM *600065). This gene codes for the β subunit of the Integrin β2 receptor family (CD18) which are exclusively expressed on the plasma membranes of neutrophil leukocytes [6]. Integrins play a key role in the margination, adherence and transmigration of neutrophil leukocytes from the blood into extravascular spaces, when stimulated to do so by inflammation-related chemotaxis gradients. Failure of this process is the hallmark of LAD. Currently four types of LAD have been well characterised. LAD II was first described in two Israeli Arab brothers, but LAD I is pan-ethnic and rare. It was first described in 1980 [7], and some hundreds of patients have been reported since then. The disease leads to severe bacterial disease, delayed separation of the umbilical stump after birth and chronic non-purulating skin and deep infections. However pyoderma gangrenosum is a rare correlate and presentation of LAD. The diagnosis of LAD is made by the demonstration of defects in random neutrophil migration and chemotaxis to various chemoattractants. Adhesion to and transmigration across endothelial cell layers are impaired. Other cell characterisation and genetic tests may be done when necessary.
Accordingly neutrophil function studies were carried out, and these (E. coli-induced migration studies and phorbol myristate acetate (PMA) activation studies) were markedly impaired compared with controls. These studies confirmed the diagnosis of LAD I. This rare disease is inherited as a Mendelian autosomal recessive and is very severe. As a recessive disorder, it is much more likely to occur when the parents are related, as were Hala’s parents. LAD carries with it a high chance of early death. The only known cure is through haematopoietic stem cell transplantation HSCT (“bone marrow transplantation”). This procedure is risky and involves ablation of the native bone marrow and its replacement with healthy donor marrow (from a family member or unrelated donor). During the procedure, which can take many weeks, the patient is at risk from severe infections and bleeding, and there is a definite associated mortality rate. The family needs to give informed consent for such a major undertaking, and this requires both parents to understand and agree. A suitable donor, preferably from the family, needs to be sought and identified. Considerable family and social support is required, and after discharge the patient remains immunocompromised for a long period requiring high-grade medical follow-up and treatment. This treatment is not available currently at Ziv Medical Centre, and unfortunately it was not possible or practicable to transfer Hala to another hospital in Israel.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


