 Viral infections can alter host immune function and contribute to bacterial superinfection, asthma exacerbations, autoimmune disease exacerbations, and reactivation of latent viruses during critical illness.
Viral infections can alter host immune function and contribute to bacterial superinfection, asthma exacerbations, autoimmune disease exacerbations, and reactivation of latent viruses during critical illness. Viral and bacterial coinfection occurs frequently in children and may have a role in complications of the viral or bacterial disease.
Viral and bacterial coinfection occurs frequently in children and may have a role in complications of the viral or bacterial disease. Viral infection can result in increased bacterial adherence to protective mucosal/epithelial barriers.
Viral infection can result in increased bacterial adherence to protective mucosal/epithelial barriers. Viral infection can alter many aspects of the innate and acquired immune response leading to vulnerability to bacterial superinfection.
Viral infection can alter many aspects of the innate and acquired immune response leading to vulnerability to bacterial superinfection. Asthmatics are more susceptible to viral infections and have heightened morbidity from exacerbations caused by viral infection.
Asthmatics are more susceptible to viral infections and have heightened morbidity from exacerbations caused by viral infection. Autoimmune disease exacerbations have been linked to several viral infections—some acute and some reactivated viruses.
Autoimmune disease exacerbations have been linked to several viral infections—some acute and some reactivated viruses. The frequency of viral infections in the pediatric population warrants consideration of the impact of viral infection on immunity in this population. For the most part, the viral illness is thought to impact on critical illness because of primary infection (bronchiolitis, croup, and influenza); however, these common infections can set the stage for bacterial coinfection that carries the risk of severe morbidity and significant mortality. Data from the recent influenza A H1N1 season highlight this association with coinfection, being not only common but also associated with more severe outcomes. Many data show that significant immune compromise can occur during critical illness of several etiologies (most notably septic shock, acute lung injury, and trauma patients not receiving immunosuppressive medications). During this time, latent viruses, such as CMV, can reactivate and potentially contribute to the severity and survival from critical illness from other etiologies.
The frequency of viral infections in the pediatric population warrants consideration of the impact of viral infection on immunity in this population. For the most part, the viral illness is thought to impact on critical illness because of primary infection (bronchiolitis, croup, and influenza); however, these common infections can set the stage for bacterial coinfection that carries the risk of severe morbidity and significant mortality. Data from the recent influenza A H1N1 season highlight this association with coinfection, being not only common but also associated with more severe outcomes. Many data show that significant immune compromise can occur during critical illness of several etiologies (most notably septic shock, acute lung injury, and trauma patients not receiving immunosuppressive medications). During this time, latent viruses, such as CMV, can reactivate and potentially contribute to the severity and survival from critical illness from other etiologies.the impact of viral infection on bronchospastic symptoms in the most severe cases.
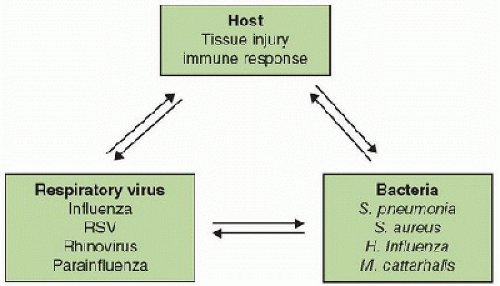 FIGURE 84.1. The interrelationship between viral infections, host immune responses, and bacteria in coinfection. Respiratory viral infection causes airway epithelial cell injury and incites an evolving immune response aimed early at eradicating virus and then tissue repair. The net effect of these responses is an anti-inflammatory milieu leaving the host vulnerable to bacterial coinfection (or subsequent infection) (1). |
 Mucous membrane surfaces, respiratory epithelium, and skin are portals of entry for many viruses. Since viral infections depend on host cellular machinery for survival and replication, entry into cells is a critical early step. This is accomplished by a variety of mechanisms, some of which are common to many viruses and others are virus specific. Examples of mechanisms shared by multiple viruses are fusion with cell membrane (enveloped viruses) and/or endocytosis after binding to cell-surface molecules. Many cell-surface molecules are exploited by viruses to gain entry into cells to facilitate viral replication. Expression of such molecules is cell-type specific and can determine tissue tropism for certain viruses. Examples of exploited host cellsurface molecules include CD4, CCR5, and CXCR4 for HIV; sialic acid residues for influenza and other respiratory viruses; heparan sulfate and proteoglycans for herpesviruses; and coxsackie adenovirus receptor (CAR), and B cell receptors CD21 and C3d for Epstein Barr Virus (2). Once in contact with cells, viral replication and the host antiviral immune response begins. Replication of lytic viruses can directly injure mucosal and epithelial protective barriers by causing cell lysis upon release of viral particles (Table 84.1). Via killing of virus-infected cells, expression of inflammatory mediators, and leukocyte infiltration, the antiviral immune response can cause further tissue injury releasing damage-associated molecules (DAMPs) that further activate the immune system (3). The immune response to microbial pathogens consists of several phases. The earliest and least specific is the innate immune response followed by a pathogen-specific adaptive immune response leading to immunologic memory. These phases of immune responses include activation of diverse cellular and humoral mediators—many of which are common to many types of infections. The immune response to viral infection involves mechanisms similar to those induced by bacterial infections as well as unique mechanisms. Recognition of viruses by the innate immune system begins by the host recognition of viral pathogen-associated molecular patterns (PAMPs) expressed during viral replication through their interaction with pattern recognition receptors (PRRs), such as the Toll-like receptors (TLRs). Viral PAMPs include virion proteins, hemagglutinin (HA), double-stranded RNA produced during replication of many viruses, F protein from RSV, single-stranded RNA, and viral DNA (4,5). TLR 2, 3, 4, 7, 8, and 9 have been implicated in responses to viral PAMPs. In fact, the initial cellular reactions to viral PAMPs are very similar to those initiated by bacterial PAMPs and are depicted in Figure 84.2 (4). The innate response is critical for recruitment of effector cells, containment of viral particles, and the initiation of adaptive or antigen-specific immune response necessary for viral eradication and immunologic memory. Binding to TLRs initiates intracellular signal transduction leading to production of proinflammatory cytokines, including type I interferons α and β (IFN-α/β), type III interferons (IFN-λ subtypes), TNF-α, IL-1β, IL-6, and chemokines, such as CCL2, CCL20, and CCR7, which facilitate trafficking of alveolar macrophages, dendritic cells (DCs), and neutrophils to sites of viral invasion (6). Anti-inflammatory cytokines, such as IL-10, soluble TNF-α receptor, and IL-1 receptor antagonist, are also induced. TLR-independent mechanisms for cytoplasmic viral detection also exist including activation of nucleotide-binding and oligomerization domain-like receptors (NLRs) and helicases including retinoic acid-inducible protein (RIG-1). NLRs are a family of multimeric cytosolic structures ultimately capable of converting pro-caspase 1 to its functional form leading to conversion of proforms of IL-1β and IL-18 to their active forms (7). A role for NLRP3 in influenza and RSV has been demonstrated, and more data are emerging to implicate other types of NLRs. The precise activators of NLRP3 are being elucidated; however, it appears that they include conventional PAMPs (dsRNA) as well as reactive oxide species and potassium (K) flux induced during viral replication (7,8,9). In contrast, RIG-1 is activated by intracellular viral dsRNA produced
Mucous membrane surfaces, respiratory epithelium, and skin are portals of entry for many viruses. Since viral infections depend on host cellular machinery for survival and replication, entry into cells is a critical early step. This is accomplished by a variety of mechanisms, some of which are common to many viruses and others are virus specific. Examples of mechanisms shared by multiple viruses are fusion with cell membrane (enveloped viruses) and/or endocytosis after binding to cell-surface molecules. Many cell-surface molecules are exploited by viruses to gain entry into cells to facilitate viral replication. Expression of such molecules is cell-type specific and can determine tissue tropism for certain viruses. Examples of exploited host cellsurface molecules include CD4, CCR5, and CXCR4 for HIV; sialic acid residues for influenza and other respiratory viruses; heparan sulfate and proteoglycans for herpesviruses; and coxsackie adenovirus receptor (CAR), and B cell receptors CD21 and C3d for Epstein Barr Virus (2). Once in contact with cells, viral replication and the host antiviral immune response begins. Replication of lytic viruses can directly injure mucosal and epithelial protective barriers by causing cell lysis upon release of viral particles (Table 84.1). Via killing of virus-infected cells, expression of inflammatory mediators, and leukocyte infiltration, the antiviral immune response can cause further tissue injury releasing damage-associated molecules (DAMPs) that further activate the immune system (3). The immune response to microbial pathogens consists of several phases. The earliest and least specific is the innate immune response followed by a pathogen-specific adaptive immune response leading to immunologic memory. These phases of immune responses include activation of diverse cellular and humoral mediators—many of which are common to many types of infections. The immune response to viral infection involves mechanisms similar to those induced by bacterial infections as well as unique mechanisms. Recognition of viruses by the innate immune system begins by the host recognition of viral pathogen-associated molecular patterns (PAMPs) expressed during viral replication through their interaction with pattern recognition receptors (PRRs), such as the Toll-like receptors (TLRs). Viral PAMPs include virion proteins, hemagglutinin (HA), double-stranded RNA produced during replication of many viruses, F protein from RSV, single-stranded RNA, and viral DNA (4,5). TLR 2, 3, 4, 7, 8, and 9 have been implicated in responses to viral PAMPs. In fact, the initial cellular reactions to viral PAMPs are very similar to those initiated by bacterial PAMPs and are depicted in Figure 84.2 (4). The innate response is critical for recruitment of effector cells, containment of viral particles, and the initiation of adaptive or antigen-specific immune response necessary for viral eradication and immunologic memory. Binding to TLRs initiates intracellular signal transduction leading to production of proinflammatory cytokines, including type I interferons α and β (IFN-α/β), type III interferons (IFN-λ subtypes), TNF-α, IL-1β, IL-6, and chemokines, such as CCL2, CCL20, and CCR7, which facilitate trafficking of alveolar macrophages, dendritic cells (DCs), and neutrophils to sites of viral invasion (6). Anti-inflammatory cytokines, such as IL-10, soluble TNF-α receptor, and IL-1 receptor antagonist, are also induced. TLR-independent mechanisms for cytoplasmic viral detection also exist including activation of nucleotide-binding and oligomerization domain-like receptors (NLRs) and helicases including retinoic acid-inducible protein (RIG-1). NLRs are a family of multimeric cytosolic structures ultimately capable of converting pro-caspase 1 to its functional form leading to conversion of proforms of IL-1β and IL-18 to their active forms (7). A role for NLRP3 in influenza and RSV has been demonstrated, and more data are emerging to implicate other types of NLRs. The precise activators of NLRP3 are being elucidated; however, it appears that they include conventional PAMPs (dsRNA) as well as reactive oxide species and potassium (K) flux induced during viral replication (7,8,9). In contrast, RIG-1 is activated by intracellular viral dsRNA produced during the replication of many viruses. Activation of IRF3 and IRF7 are critical for induction of type 1 interferons (IFN-α /β) that are essential antiviral cytokines (10). These pleiotropic cytokines can affect viral replication by reducing “viral receptor” molecule expression on the host cell surface, inhibition of transcription and translation of viral proteins through IFNα /β-stimulated genes such as MxA protein, 2′5′-oligoadenylsynthase, double-stranded RNA kinase (PKR), and eukaryotic initiation factor α (eIF-2α), thereby inhibiting viral replication (5,6). In addition to viral proteins, RNA/DNA-mediated activation of the above pathways and DAMPs released from injured tissue including lysed epithelial cell contents can activate innate immune pathways via their respective TLR, RLR, or inflammasomes (10).
TABLE 84.1 MECHANISMS OF BACTERIAL ADHERENCE TO HOST CELLS DURING VIRAL INFECTION | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
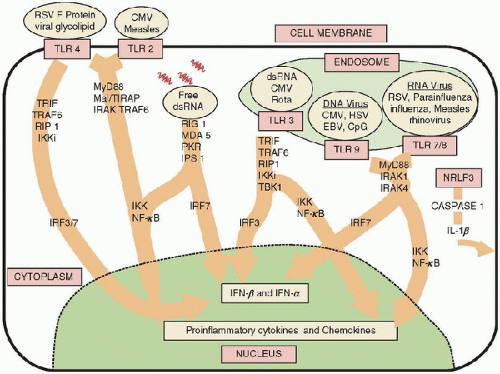 FIGURE 84.2. Early signaling initiated by viral PAMPs. Many viruses are recognized by TLRs. Viral PAMPs also include the fusion protein (F protein) of RSV, dsRNA, DNA, HA protein, and envelope proteins. The PAMPs associated with many viruses capable of signaling through TLRs have not been identified. Intracellularly, many of the adaptor molecules and kinases activated by TLRs are shared and, as with bacteria, ultimately initiate similar inflammatory cascades via NF-κB- and IFN-β-mediated signaling. Both of these pathways are important in containment of viral infections. Since many aspects of the signaling pathways activated by both viral and bacterial PAMPs are shared, the impact of sequential signaling (e.g., viral followed by bacterial) has not been well characterized (4). Rota, rotavirus; CpG, cytosine-phosphate-guanine sites. |
 Elegant data have demonstrated that during respiratory viral infection there is disruption of protective epithelial barriers as a result of diversion of cell machinery to intracellular viral replication often with direct lysis of infected epithelial cells (18). When protective epithelial barriers are disrupted, mucociliary dysfunction can occur as well as exaggerated bacterial colonization by adherence to exposed basement membrane elements permitting penetration into deeper tissues (see Table 84.1). This concept was demonstrated in experimental human inoculation with influenza followed by the development of detectable airway colonization with Streptococcus pneumoniae 6 days after inoculation with influenza (23). In addition, several studies have demonstrated significantly increased nasopharyngeal colonization with commensal organisms during other respiratory
Elegant data have demonstrated that during respiratory viral infection there is disruption of protective epithelial barriers as a result of diversion of cell machinery to intracellular viral replication often with direct lysis of infected epithelial cells (18). When protective epithelial barriers are disrupted, mucociliary dysfunction can occur as well as exaggerated bacterial colonization by adherence to exposed basement membrane elements permitting penetration into deeper tissues (see Table 84.1). This concept was demonstrated in experimental human inoculation with influenza followed by the development of detectable airway colonization with Streptococcus pneumoniae 6 days after inoculation with influenza (23). In addition, several studies have demonstrated significantly increased nasopharyngeal colonization with commensal organisms during other respiratory viral infections (parainfluenza, RSV, and adenovirus) when compared to baseline colonization in the same children (23). Specifically, bacterial adherence is enhanced in areas where epithelium has been denuded. This has been seen in many experimental settings with cell culture and in vivo animal models. In addition, during the 1957 influenza epidemic at autopsy, direct visualization of Staphylococcus aureus in the lungs adherent to areas of denuded tracheobronchial epithelium was seen (18,24,25).
TABLE 84.2 IMMUNOMODULATORY EFFECTS OF VIRAL INFECTIONS | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|




