 The immune system is a phenomenally complex biologic system.
The immune system is a phenomenally complex biologic system. Critical illness (both septic and nonseptic) elicits an immune response that is designed to protect the body.
Critical illness (both septic and nonseptic) elicits an immune response that is designed to protect the body. An unbalanced immune response may worsen the overall clinical state and exacerbate the level of organ dysfunction.
An unbalanced immune response may worsen the overall clinical state and exacerbate the level of organ dysfunction. Many examples of congenital immunodeficiency provide the clinician with important insight into infection susceptibility, mechanisms of systemic inflammation, and multiorgan dysfunction.
Many examples of congenital immunodeficiency provide the clinician with important insight into infection susceptibility, mechanisms of systemic inflammation, and multiorgan dysfunction. To date, no interventional trial of immune modulation in critically ill children has been of proven benefit, perhaps because of a failure of appropriate targeting.
To date, no interventional trial of immune modulation in critically ill children has been of proven benefit, perhaps because of a failure of appropriate targeting. Future clinical trials of immunomodulative agents must evaluate the patient’s immunocompetence and inflammatory and infectious status as well as be rigorously tested for in vivo efficacy.
Future clinical trials of immunomodulative agents must evaluate the patient’s immunocompetence and inflammatory and infectious status as well as be rigorously tested for in vivo efficacy. Therapeutic trials should be appropriately targeted to the immune state of the individual.
Therapeutic trials should be appropriately targeted to the immune state of the individual.TABLE 81.1 CHARACTERISTICS OF THE INNATE VERSUS ADAPTIVE IMMUNE SYSTEM | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 81.2 SEVERE COMBINED IMMUNODEFICIENCY SYNDROMES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
severe combined immunodeficiency disease [SCID; Chapter 86], opportunistic infections [Chapter 95], Epstein-Barr virus (EBV) driven lymphoproliferative disease [Chapter 104], tumor lysis syndrome and neutropenic fever [Chapter 115], and idiopathic pneumonitis syndrome, post-bone marrow transplant respiratory failure, and veno-occlusive disease [Chapter 117]). In addition to these severe defects, a number of conditions have been recognized, many originating from single-gene deletions or polymorphisms, that are providing valuable insights into the complex processes of inflammation. Immunodeficiencies are listed in Tables 81.2 and 81.3.
TABLE 81.3 OTHER CONGENITAL IMMUNODEFICIENCY SYNDROMES | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
process of immune restoration following stem cell transplantation, or in those with Cryptosporidium infections and CD40L deficiency. The relationship between infectious susceptibility and severity of the ensuing inflammatory response is complex and frequently determines the clinical course of critically ill patients. A number of defects in the immune system highlight the balance between risk of infection and severity of the host response. Five immune networks have been chosen to illustrate this point: complement system, MBL, endotoxin recognition, cytokines and the inflammasome.
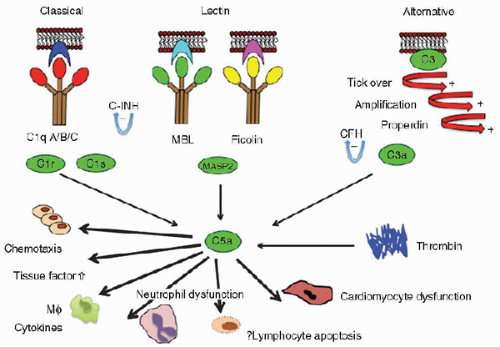 FIGURE 81.1. The complement and lectin pathways. The lectin pathway of complement is activated by MBL and ficolins. On binding to appropriate targets, MBL-MASP-2 complexes cleave C4 and C2 to form C3 convertase (C4bC2a). MBL-MASP-1 complexes may activate C3 directly. Ficolins also work in combination with the MASPs. The classic and alternative pathways also generate C3 convertase enzymes, which cleave C3. The lytic pathway (C5-C9) is common to all three routes of C3 cleavage. MASP, MBL-associated serine proteases; MASP-1, MBL-associated serine protease-1; MASP-2, MBL-associated serine protease 2. C-INH is C1 esterase inhibitor; CFH is Complement Factor H. |
be influential in this environment. Children who are admitted to intensive care following infection, trauma, or surgery have a greatly increased risk of developing the systemic inflammatory response syndrome (SIRS) within the first 48 hours of PICU admission if they have MBL deficiency (odds ratio [OR], 7.1; 95% confidence interval [CI], 2.9-19; p = 0.0001). This is true for both infectious (OR, 11; 95% CI, 2-57; p = 0.001) and noninfectious illness (OR, 5; 95% CI, 1.5-19; p
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


