Temperature (°C)
Cerebral metabolic rate (% of baseline)
Safe duration of HCA (min)
37
100
5
30
56 (53–60)
9 (8–10)
25
37 (33–42)
14 (12–15)
20
24 (21–29)
21 (17–24)
15
16 (13–20)
32 (25–38)
10
11 (8–14)
45 (36–62)
A second mechanism for the protective effect of hypothermia is through depression of presynaptic release of excitatory amino acid transmitters (e.g., glutamate and aspartate). This leads to a decrease in excitotoxicity and a subsequent decrease in the extent of neuronal damage after circulatory arrest [32, 44, 45].
Finally, in a study where swine were cooled to 19 °C and placed in circulatory arrest for 90 min and histologically examined at various times from 1 h to 1 week after circulatory arrest, neuronal damage was most significant in the animals that were histologically examined 8–72 h after circulatory arrest. They noted that caspace-3 and -8, indicators of apoptosis, were elevated. Caspace-3 remained elevated for 72 h [19]. Increased activity was also seen in cytosolic cytochrome c . and fos-protein. Apoptotic histological changes were observed in central nervous system neurons. The authors concluded that apoptosis is one of the pathological processes involved with delayed neurological deficits following hypothermic circulatory arrest.
With few exceptions, pharmacological protective strategies have not been found to protect the human central nervous system from ischemic damage during cardiopulmonary procedures [46–61] (Table 39.2). Hence, we depend on deep hypothermia as one of our primary techniques for brain protection.
Table 39.2
Randomized, placebo-controlled trials of pharmacologic neuroprotection for adults undergoing cardiac surgery
Drug | Proposed primary mechanism | References | n | Type of surgery | Main findings |
|---|---|---|---|---|---|
Thiopental | ↓ CMRO2 | Nussmeier et al. [46] | 182 | Valvular | Thiopental ↓ cognitive complications 10 days after surgery |
Zaidan et al. [47] | 300 | No difference in neurologic outcomes thiopental versus placebo | |||
Propofol | ↓ CMRO2 | Roach et al. [48] | 300 | CABG | No difference in cognitive complications 5–7 days or 50–70 days after surgery propofol vs. controls |
Nimodipine | Ca++channel blocker | Legault et al. [49] | 225 | Valvular | Study terminated early due to higher mortality in treated vs control group; no evidence of benefit with nimodipine on cognitive outcomes |
Prostacyclin | ↓ platelet aggregation, ↓ inflammation | Fish et al. [50] | 150 | Valvular | No difference in cognitive outcomes 2 weeks after surgery between treated and control patients |
Aprotinin | Mechanism(s) unknown; may be due to ↓↓ pericardial aspirate | Levy et al. [54] | 287 | CABG | No strokes in “high” and “low” dose aprotinin groups vs controls (n = 5) and “pump” prime only (n = 1) groups (P = 0.01) inflammation |
Harmon et al. [55] | 36 | CABG | Cognitive deficits 2 weeks after surgery lower in aprotinin vs placebo group (23 vs. 55 %, P<0.05) | ||
Lidocaine | Na + channel blockade; membrane stabilization/↓ EAA release | Mitchell et al. [56] | 55 | Valvular | Neurocognitive outcome better 10 days and 10 weeks after surgery in lidocaine vs placebo group but not at 6 months |
Wang et al. [57] | 42 | CABG | Improved neurocognitive function 9 days after surgery with lidocaine vs. placebo | ||
Clomethiazole | GABA receptor agonist | Kong et al. [58] | 219 | CABG | No difference in neurocognitive function 4–7 weeks after surgery in clomethiazone vs placebo groups |
Pexelizumab | ↓ C5a and C5b-9 | Mathew et al. [59] | 800 | CABG | Pexelizumab had no effect on global cognition but did lower decline in the visuo-spatial domain compared with placebo |
The protection offered by deep hypothermia, however, is limited to relatively short periods of time (see Table 39.1) [34]. Hence, selective antegrade cerebral perfusion is often administered as an adjunct protective scheme during the surgical procedure [62–75].
During the rewarming phase following circulatory arrest, CMR02 increases along with an increase in cerebral blood flow. Typically, in the absence of selective antegrade cerebral perfusion, there is an initial cerebral blood flow hyperemia, which is caused by an oxygen debt accumulated during the arrest phase [29, 30, 35, 37, 38]. This reflects residual metabolic activity present even at profound hypothermic brain temperatures. After a short hyperemic phase, cerebral blood flow decreases inappropriately for the increased metabolic demand present during and after rewarming. Thus, cerebral blood flow again becomes uncoupled from metabolic demand [29, 30, 37–39]. There is evidence of increased production (i.e., up regulation) of endothelial adhesion molecules, which tend to trap “clusters” of leukocytes in the small cerebral blood vessels. This leads to leukocyte activation with subsequent blocking of cerebral blood vessels and an increase in the postoperative inflammatory process [29, 30, 35, 37]. Webster et al. [76] recently demonstrated that, during global cerebral ischemia, hypothermia decreased the activation of DNA-bound NF-Kappa B. NF-Kappa B . is a major transcription factor regulating genes associated with inflammatory mediators. Inhibition of this transcription factor attenuates inflammation.
Another occasional consequence of circulatory arrest is increased intracranial pressure caused by cerebral edema [77]. This develops after cardiopulmonary bypass is reinstituted. It can be detected by observing bulging fontanelles in neonates or infants. If a transcranial Doppler is available, a decrease or reversal of diastolic cerebral blood flow velocity is detected [78]. The increased intracranial pressure can be reversed by 10–15 min of cold perfusion after cardiopulmonary bypass is resumed following a period of circulatory arrest [77].
Temperature Monitoring
Because of the importance of profound hypothermia for protecting the central nervous system, many surgeons rely on body temperature to decide when an appropriate decreased target brain temperature has been reached prior to beginning circulatory arrest. Direct brain temperature is usually not available during routine cardiac procedures. Clinicians must rely on extracerebral temperature monitoring for determining a safe temperature for initiating circulatory arrest. Stone et al. [79] were the first to compare the accuracy of several surrogate temperature monitoring sites to temperatures acquired directly from the brain. Their patients were undergoing open craniotomies for clipping of giant aneurysms, which required periods of circulatory arrest. Temperature probes were placed 4 cm within the depth of the cerebral cortex and were compared to temperature data recorded from the bladder, rectum, pulmonary artery, tympanic membrane, nasopharynx, esophagus, and skin of the axilla and lower limbs. Comparisons were made during cooling to 16 °C and during rewarming to 37 °C after a period of circulatory arrest. Prior to cooling, with the exception of the sole of the foot, temperatures from all recording sites were equal. During rapid cooling in preparation for circulatory arrest, none of the surrogate monitoring sites tracked the brain’s temperature well (Fig. 39.1). All temperatures lagged behind the brain temperature. The nasopharyngeal and esophageal temperature probes showed the smallest difference in comparison to direct brain temperatures. Just prior to initiating circulatory arrest, the difference between the temperatures at the surrogate sites and the actual brain temperature was ± 2.8 °C. During rewarming, the same phenomenon was noted. Here, nasopharyngeal and esophageal temperature probes recorded temperatures that were closest to the brain temperature. Of concern, when the nasopharyngeal temperature reached 37 °C, the brain was typically 1–2 °C higher. As hyperthermia is known to increase the degree of ischemia-induced brain damage, these results were concerning [80–85]. The variability across patients, as well as across temperature-monitoring sites, precluded choosing one site as the best surrogate temperature for predicting brain temperatures during rapid temperature changes. Stone et al. [79] also studied patients undergoing excision of brain tumors during normothermic conditions. Temperatures recorded at cortical depths of 1 cm were typically cooler by 0.5–3 °C compared to temperature probes placed at cortical depths of 4 cm. In addition, the superficial cortical temperature recordings showed greater variability, particularly when cold irrigation fluids were used. They reasoned that the variability of the superficial temperature recordings was caused by the effects of ambient temperature and cool irrigation fluids.
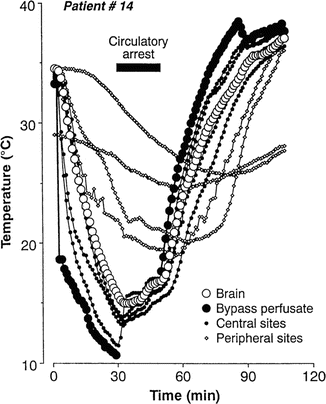
Fig. 39.1
Plots of temperature changes during rapid cooling and rewarming of patients on cardiopulmonary bypass (CPB) . Temperatures acquired from the brain, CPB arterial inflow (bypass perfusate), central sites (i.e., pulmonary artery, nasopharynx, esophagus, tympanic membrane), and peripheral sites (bladder, rectum, axilla, sole of foot). With the exception of the sole of the foot, all temperatures were equal prior to cooling. Sites which tracked the brain temperatures the closest were the nasopharynx and esophagus. Reprinted with permission from Stone et al. [79]
Crowder et al. [86] subsequently studied the correlation of brain temperature to tympanic membrane, esophagus, bladder, pulmonary artery, and the jugular bulb temperature. The jugular bulb was considered to be an excellent monitoring site as it drained 60–66 % of venous blood flow from the homologous cortex, with 1 % contamination from extracranial sources [86–88]. Brain temperatures were monitored with temperature probes placed in the subdural space distant to the craniotomy site (i.e., under the intact cranium , so as to minimize the effects of ambient room temperature on the subdural temperature recordings). Moderate hypothermia was induced with the lowest reported brain surface temperature reaching 32.3 °C. Their results confirmed Stone’s study and showed that large fluctuations of the brain surface temperature (i.e., subdural temperature) caused by irrigation fluid and ambient temperature made the comparison of jugular bulb and true parenchymal brain temperature unreliable. Additionally, jugular bulb temperatures tended to be higher than all other monitoring sites during rewarming. Nevertheless, studies following Crowder et al.’s report have assumed that the jugular bulb can be used to track brain temperature during rapid temperature changes. Grocott et al. [87] showed that nasopharyngeal temperatures were lower than jugular bulb temperatures, but, in general, they tracked jugular bulb changes accurately during rapid cooling. At rewarming, however, jugular bulb temperatures were consistently 1–2 °C higher (implying that brain temperature may be higher than nasopharyngeal temperature). Similar results have been reported by other investigators [88].
In summary, all investigations have demonstrated that surrogate temperature data tend to lag behind true brain temperatures during rapid temperature changes. The greatest bias is found during rewarming and can lead to brain temperature reaching 1–3 °C higher than recorded from surrogate temperature sites [79]. A second conclusion derived from all investigations is that the variability across patients, as well as the data from surrogate temperature sites, precludes choosing one best site for accurately predicting brain temperatures during rapid cooling and rewarming.
Neuroelectrophysiological Monitoring
The appropriate temperature for initiating circulatory arrest should be cold enough for reducing adverse neurological outcomes through the mechanisms discussed in the section on “Temperature Monitoring.” On the other hand, excessive cooling may lead to adverse outcomes secondary to intraneuronal ice crystal formation with subsequent injury (e.g., usually expected at temperatures below 10 °C) [40]. Secondly, longer bypass time is required in order to achieve these low temperatures as well as increased time for rewarming. Hence, several investigators have studied the use of temperature-related changes of the electroencephalogram (EEG) and somatosensory-evoked potentials (SSEPs) for determining the appropriate temperature for initiating circulatory arrest [40, 41].
Stecker et al. studied typical changes seen in the EEG during cooling prior to circulatory arrest (Fig. 39.2). Four EEG patterns were noted (please see Chap. 10 for review of EEG monitoring). The order of appearance with decreasing body temperatures is as follows: pre-cooling EEG (Fig. 39.2d), appearance of periodic complexes (Fig. 39.2e), burst suppression (Fig. 39.2f), and electrocerebral silence (Fig. 39.2g). The distribution of patients demonstrating each of the characteristic EEG patterns as a function of temperature is shown in Fig. 39.2a–c. The importance of these results is that there is a range of temperatures rather than a single best temperature for which each of the sequential EEG patterns occurs during cooling on bypass. The mean nasopharyngeal temperature where periodic EEG complexes are noted was 29.6 ± 3 °C. Burst suppression occurred at 24.4 ± 4 °C and electrocerebral silence at 17.8 ± 4 °C. Therefore, typically the patients will be cooled at 2 °C below the temperature at which suppressed EEG is achieved prior to initiating circulatory arrest.
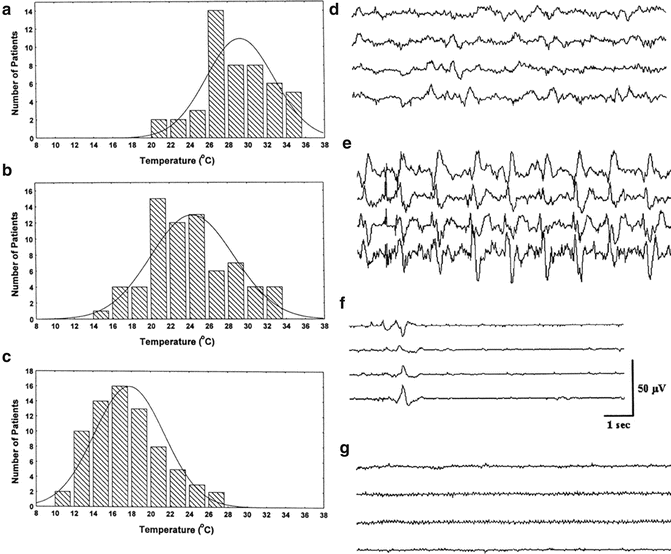
Fig. 39.2
The distribution of nasopharyngeal temperatures for patients developing (a) periodic EEG samples, (b) burst suppression, and (c) electrocerebral silence during rapid cooling prior to circulatory arrest. Examples of typical EEG patterns prior to cooling are shown in (d): Periodic complexes and burst suppression are shown in (e) and (f), respectively. (g) shows an example of EEG suppression. Reprinted with permission from Stecker [93]
Please refer to Chap. 1 for an in-depth review of SSEP responses. Figure 39.3 demonstrates the relationship between the cortical (N20), spinal cord (N13), and Erb’s point (i.e., brachial plexus) response latencies with temperature [88]. Note that latency increases at each recording site as body temperature decreases. This temperature effect on latency is due, in part, to the effect of temperature on conduction velocity along axons such that, at 2.7 and 7.2 °C, conduction blocks occur in unmyelinated and myelinated axons, respectively [88]. These effects are in turn due to the effects of hypothermia on the rate of change of sodium channels within axonal membranes [89, 90]. Stecker demonstrated an increase in SSEP refractory periods as body temperature decreases, suggesting that a slower stimulation frequency and increased stimulation intensity are required for acquiring SSEP responses at colder temperatures [40, 90, 91].
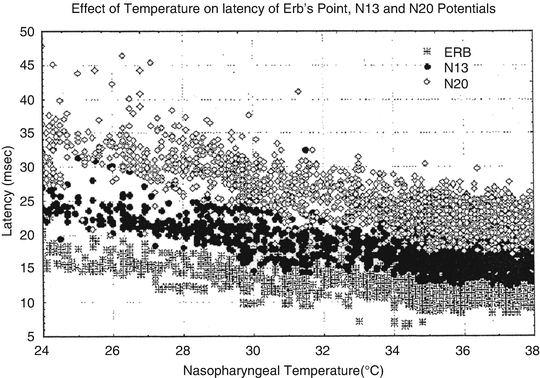
Fig. 39.3
This figure shows the differential sensitivity to cooling of the latencies for the N20 (sensory cortex response), N13 (spinal cord dorsal column responses near the brainstem), and ERB (brachial plexus activation). Note the limited change in latency of the brachial plexus responses with increasing prolongation of the spinal cord dorsal columns and sensory cortex latencies with colder temperatures. Reprinted with permission from Stecker [91]
Figure 39.4 demonstrates that at sufficiently low body temperatures, SSEPs are lost. This phenomenon offers a second physiological parameter, in addition to EEG changes, for determining an appropriate target temperature for suppressing electrical activity in the brain. Figure 39.5 demonstrates the orderly disappearance of the thalamocortical (C) , followed by the suppression of the cervicomedullary responses (D, E) as colder nasopharyngeal temperatures are reached. Figure 39.5a, b shows the distribution of nasopharyngeal temperatures for loss of the cortical and cervicomedullary responses. The N20-P22 complex acquired from thalamic and cortical generators disappears with body cooling to a nasopharyngeal temperature of 21.4 ± 4 °C and the cervical medullary responses (N13) are lost at 17.8 ± 4 °C. These SSEP values are consistent with the results acquired by Ghariani et al. [92]. As was the case for EEG changes during cooling, a single targeted temperature is inadequate for accurately predicting the individual brain and spinal cord electrical suppression during induced hypothermia. EEG, cortical, and subcortical SSEP responses (for determining subcortical depression) were monitored during the cooling phase of our case. The results were used for determining the appropriate nasopharyngeal temperature for initiating circulatory arrest.
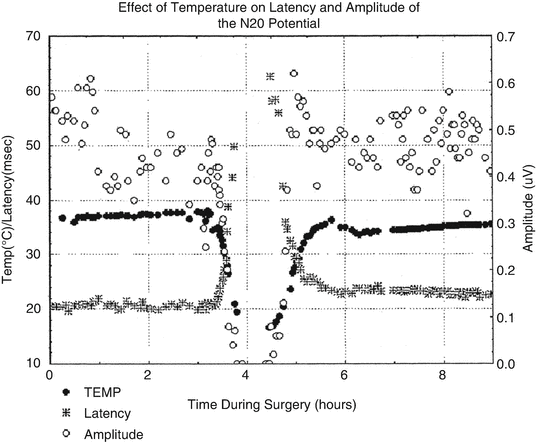
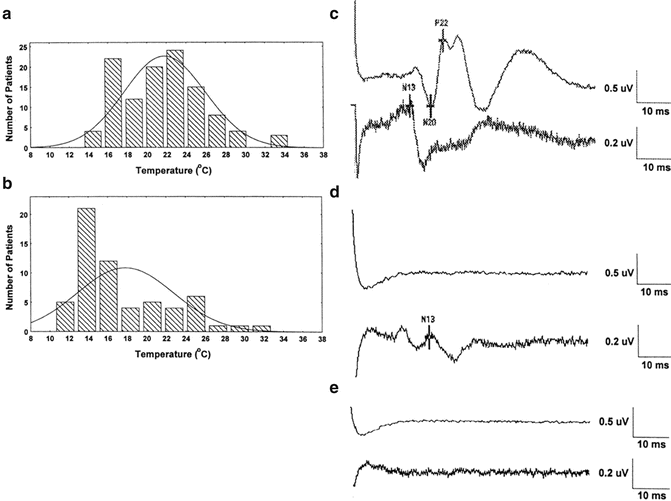
Fig. 39.4
The effect of cooling and rewarming of the somatosensory cortex N20 latency and amplitude. The variability of the N20 amplitude is greater than the latency. The N20 latency is probably a better N20 parameter for monitoring the effect of temperature on the cortex. Reprinted with permission from Stecker et al. [91]
Fig. 39.5
The distribution of nasopharyngeal temperatures for loss of the N20-P22 cortical SSEP complex (a) and spinal cord N13 responses (b) in patients undergoing rapid cooling prior to circulatory arrest. Examples of typical SSEPs during rapid cooling prior to cooling (c), after loss of the cortical SSEP responses with prolongation of the N13 (d) and loss of the N13 spinal cord response. Reprinted with permission from Stecker et al. [93]
After the portion of the surgical repair of the patient’s dissection is carried out during circulatory arrest and selective antegrade cerebral perfusion, the patient is rewarmed. Electrophysiologic modalities for tracking the rewarming process have also been studied [93]. During this phase of the procedure, the EEG and SSEP responses take on an additional significance beyond merely tracking the rewarming effects on the central nervous system. In this case, they can also predict the occurrence of new neurological deficits occurring during the circulatory arrest phase of the procedure.
Stecker demonstrated the pattern of EEG and SSEP responses observed during rewarming prior to separation from bypass. In neurologically normal patients, the order of reappearance of SSEP and EEG activity was consistent across the patients studied. The N13 SSEP waveform reappeared first after rewarming followed by the thalamocortical SSEP responses (i.e., N20-P22). EEG burst suppression activity appeared next followed by continuous EEG activity, which occurs on average 28.1 min later. The rewarming rate on average was 0.42 ± 0.19 °C per minute from a circulatory arrest nasopharyngeal temperature of 14.4 ± 2 °C. In general, the time of recovery of the N20-P22 SSEP responses during rewarming increased by 0.3–1.0 min for each minute of circulatory arrest. Of importance, the time as well as the nasopharyngeal temperature for reappearance of continuous EEG and the thalamocortical SSEP (i.e., N20-P22) responses were significantly longer and higher, respectively, in patients who developed new postoperative neurological deficits compared with patients with normal postoperative neurological outcomes (Tables 39.3 and 39.4). Based on these results, a multivariable analysis allowed calculation of the relative risk of new postoperative deficits occurring during the circulatory arrest and initial rewarming phases of the operation by determining the product of 1.56 and 1.27 and the temperature for the reappearance of continuous EEG and N20-P22 SSEP responses, respectively (see Table 39.5). This approach accurately predicted new postoperative neurological deficits 89 % of the time. Ghariani et al. [92] found similar delays in recovery of central nervous system (i.e., CNS) electrical activity in patients with new postoperative neurological deficits [92].
Table 39.3
Summary of times and temperature for events during rewarming in patients neurologically intact postoperativelya
Description | Factor | n | Mean (SD) | Factor | Mean, SD | Factor | Mean (SD) |
|---|---|---|---|---|---|---|---|
Reappearance of burst suppression | TRas | 48 | 19.0 (9) | NTRas | 21.2 (5) | CTRas | 20.6 (3) |
Reappearance of continuous EEG | TRCont | 47 | 47.1 (26) | NTRCont | 30.1 (5) | CTRCont | 26.8 (4) |
Reappearance of N20-P22 | TRN20 | 66 | 14/2 (7) | NTRN20 | 18.6 (3) | CTRN20 | 20.0 (3) |
Reappearance of N13 | TRN13 | 37 | 12.6 (6) | NTRN13 | 17.2 (2) | CTRN13 | 19.2 (2) |
Table 39.4
Summary of times and temperature for events during rewarming in patients with neurological deficits a
Description | Factor | No. | Mean (SD) | P b | Factor | Mean (SD) | P | Factor | Mean (SD) | P |
|---|---|---|---|---|---|---|---|---|---|---|
Reappearance of burst suppression | TRas | 9 | 26.2 | 0.02 | NTRas | 24.8 (6) | 0.07 | CTRas | 22.5 (5) | 0.23 |
Reappearance of continuous EEG | TRCont | 9 | 80.5 (28) | 0.0008 | NTRCont | 36.2 (0.8) | 0.0007 | CTRCont | 32.8 (4) | 0.0006 |
Reappearance of N20-P22 | TRN20 | 16 | 20.3 (9) | 0.0004 | NTRN20 | 22.3 (4) | 0.0002 | CTRN20 | 20.0 (2) | 0.95 |
Reappearance of N13 | TRN13 | 10 | 16.5 (9) | 0.12 | NTRN13 | 18.8 (2) | 0.07 | CTRN13 | 19.7 (3) | 0.45 |
Table 39.5
Multivariable analysis of factors associated with either intraoperative stroke or postoperative confusion by stepwise logistic regressiona
Factor | Relative risk (per °C) | 95 % CI | P value |
|---|---|---|---|
NTRCont | 1.56 | 1.1–2.2 | <0.001 |
NTRN20 | 1.27 | 1.02–1.56 | 0.015 |
Finally, other investigators demonstrated that loss of SSEP responses prior to cooling or following circulatory arrest during aortic surgery correlated with new deficits [92, 94, 95]. Cheung et al. [94], however, demonstrated a large degree of variance for cortical SSEP amplitudes acquired from patients with normal outcomes following bypass procedures. In order to predict new focal deficits, a 21-fold left–right amplitude asymmetry of the N20-P22 (i.e., thalamocortical) response was required [94].
Acid Base Management on Bypass
Hypothermia has profound effects on neutral pH of water. During cooling, CO2 solubility increases, shifting the equilibrium of the following equation to the right.
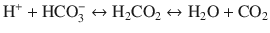 This shift results in an increase in pH during cooling.
This shift results in an increase in pH during cooling.
There are two management approaches for dealing with these arterial blood gas results. The first is referred to as the pH stat management scheme where the pH is kept constant. In this case, the perfusionist will either add CO2 to the patient’s blood or reduce the gas flow entering the oxygenator. This is done to correct the hypothermic blood pH to 7.4, at the expense of an increased load of CO2. Increased CO2 leads to vasodilatation of the cerebral arterioles [29, 35, 96–99]. This uncouples autoregulation prior to 21–22 °C (i.e., where it is lost during alpha stat management). Cerebral blood flow increases in the cerebral cortex, as well as the brainstem, leading to a decrease in temperature gradients. Temperature gradients might exist when pH stat management is not used during rapid cooling.
The second management scheme is the alpha stat management where the state of intracellular charges is kept constant. In this case, blood gas pH is not corrected for temperature. Thus, the perfusionist does not add CO2 to the blood. Autoregulation is maintained for a longer period of time during cooling, with the expected decrease in cerebral blood. It is interesting to note in this regard that functioning poikilotherms use the alpha stat blood gas management. On the other hand, hibernating animals use pH stat [29, 30, 37]. Perhaps the reason for these preferences lies in the fact that pH stat management leads to a more uniform hypothermic body temperature and perhaps improved protection during hibernation.
Neonates, when cooled using alpha stat management for circulatory arrest, had an elevated incidence of choreoathetosis [96, 98, 100]. The incidence of this postoperative deficit decreased when pH stat management was used. This suggested improved and uniform hypothermic protection of the brainstem due to increased vasodilation. Animal studies showed an increased tolerance to circulatory arrest when pH stat management was used [100]. Furthermore, the alkalotic pH obtained with alpha stat management in conjunction with hypothermia shifts the oxyhemoglobin saturation curve to the left. This reflects increased affinity of hemoglobin for oxygen during conditions of high pH and low temperature [96–100]. Thus, high venous oxygen saturations with alpha stat management during cooling may not be caused by a cold brain extracting less oxygen from hemoglobin , but because hemoglobin will not release oxygen under cold, alpha stat management [96, 100]. With pH stat, on the other hand, left shift of the oxyhemoglobin saturation will decrease due to the increased acidity in the hypothermic patient’s blood [100]. This implies that more oxygen will be available to brain cells during hypothermic circulatory arrest than with alpha stat management. However, oxygen supplies to the hypothermic brain during circulatory arrest may be principally met by dissolved oxygen in the stagnant cerebral blood volume during circulatory arrest [33, 35, 38]. In any event, caution should be exercised when interpreting increased venous oxygen saturations during hypothermia as a reflection of decreased oxygen extraction due to cold-induced reductions in metabolic oxygen demand when alpha stat management is used [98, 100].
Related posts:
 Deep Brain Stimulation
Deep Brain Stimulation
 Intraoperative Neurophysiologic Monitoring During Surgery for Supratentorial Mass Lesions
Intraoperative Neurophysiologic Monitoring During Surgery for Supratentorial Mass Lesions
 Surgery for Chiari Type I Malformation
Surgery for Chiari Type I Malformation
 Intraoperative Neurophysiological Monitoring for Intracranial Aneurysm Surgery
Intraoperative Neurophysiological Monitoring for Intracranial Aneurysm Surgery
 Trigeminal Microvascular Decompression
Trigeminal Microvascular Decompression
 Monitoring of Jugular Venous Oxygen Saturation
Monitoring of Jugular Venous Oxygen Saturation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


