Support of the Failing Circulation
KEY POINTS
1 Circulatory insufficiency and shock result from inadequate perfusion relative to tissue demands. Although certain physical and laboratory parameters may be suggestive, shock is defined by overt dysfunction of key vital organs—not by parameters that selectively reflect either oxygen supply or demand.
2 Attention to the demand side of the perfusion imbalance is a potent and often overlooked means of reversing the pathophysiology of shock.
3 Early goal-oriented intervention may be crucial in deciding outcome.
4 Three basic mechanisms may cause or contribute to circulatory insufficiency: pump failure, insufficient vascular tone (vasoplegia), and hypovolemia. Heart rate and rhythm as well as the determinants of stroke volume (preload, contractility, and afterload) should be considered independently for their potential to contribute to cardiovascular dysfunction.
5 The parameters that characterize heart function must be scaled to body size; any specific value of cardiac output oxygen consumption, or vascular resistance may take on different significance for large and small patients.
6 During shock, the respiratory muscles may outstrip the heart’s ability to deliver adequate blood flow to them. Mechanical support may relieve the ventilatory burden, thereby increasing the blood flow available to other marginally perfused organs.
7 Repeated examination of mental status, urine output, and skin perfusion provides information essential in guiding therapy. Invasive monitoring with arterial and pulmonary artery catheters, in conjunction with the fluid challenge, provide the data necessary to wisely select vasoactive agents and regulate the rates of volume and drug infusion.
8 Adequate circulatory volume must be ensured before vasopressors are used. The type of fluid should be selected by considering the need for blood, the nature of the fluid lost from the vascular space, the acuity of the problem, the urgency of reversal, the financial cost, and the potential risk of the product to the patient.
9 Most (but not all) vasoactive agents used to support the circulation are catecholamine derivatives with α, β1, or β2 activity, in varying proportions. The relative intensity of each effect may vary with dosage.
10 Mechanical interventions (ventilatory support, positive end-expiratory pressure, aortic balloon pumping) may be needed to reduce afterload or modify preload. Once initiated, these interventions should be maintained only as long as necessary but should be withdrawn cautiously.
▪ PHYSIOLOGY OF THE FAILING CIRCULATION
Circulatory Insufficiency
Circulatory insufficiency and shock reflect inadequate perfusion relative to tissue demands. Although certain physical and laboratory parameters may be suggestive, shock is defined by overt dysfunction of vital organ systems—not uniquely by such “supply side” parameters as blood pressure (BP) or cardiac output (CO) or by such “demand side” parameters as oxygen consumption ([V with dot above]O2). What might be considered a normal CO in a healthy patient at rest may inadequately perfuse the tissue beds of a critically ill patient. The prime objective of circulatory support, therefore, is to maintain nearoptimal vital organ perfusion, as reflected in mental status, urinary output, and systemic pH, at acceptable cardiac filling pressures.
Organ perfusion is governed by driving pressure and vascular resistance. Ordinarily, an adequate pressure gradient is present, and vasomotor tone regulates individual organ perfusion in proportion to metabolic demand. Under resting conditions, only a small percentage of all vascular channels are fully open. However, when the available pressure fails to maintain adequate flow despite optimized vasodilation (e.g., during a cardiovascular crisis or hypovolemia) or when defective vasoregulation fails to maintain perfusion pressure or flow distribution (e.g., during sepsis), vital tissues are not adequately nourished to maintain all normal cellular functions. The vascular beds of different organs vary in the extent to which they can compensate for deprivation of normal regional blood flow and/or drop in circulatory pressure. Certain conditions, such as sepsis, may interfere with these delicate vasomotor controls. The shock syndrome may be initiated when these compensatory mechanisms reach their limits. Once shock physiology is under way, vasoactive mediators and products of inflammation, some with myocardial depressant properties, may be released into the circulation system to perpetuate the circulatory crisis. Even with appropriate treatment, mortality exceeds 50% for septic shock and for cardiogenic shock unaddressed by coronary reperfusion.
Determinants of Cardiac Output
Although attention usually is focused on the pump that energizes the circulation (the heart), vascular compliance and tone are potentially of equal importance. Thus, whereas the Frank-Starling relationship offers a useful if somewhat restricted perspective on circulatory kinetics, CO can be viewed equally well as a function of the effective pressure gradient driving blood from the periphery back to the heart and the resistance to venous return. The average upstream peripheral force driving venous return, the mean systemic pressure (MSP), is the equilibrium pressure that would exist throughout the vasculature if the heart abruptly stopped pumping. Because of the large capacitance of the venous relative to the arterial bed, MSP (normally 7 to 10 mm Hg) lies much closer in value to the central venous pressure (CVP) than to mean arterial pressure (MAP). MSP is influenced by both blood volume and vascular tone. Early in many shock states, aggressive filling of the vasculature is often required to compensate for the vasoplegia that otherwise would cause MSP to fall. Capacitance beds must be filled before vessel tone and MSP can rise to adequate levels. (Vigorous early resuscitation of the intravascular compartment helps account for the reported success of “goal-directed therapy.”) The downstream back pressure to venous return is right atrial pressure (PRA). If MSP fails to rise sufficiently to compensate for an increase in PRA, CO falls. Indeed, the relationship between CO and PRA is linear for any fixed value of MSP, and the slope of this relationship is influenced by the resistance to venous return. The tendency of the vena cava to collapse limits the extent to which effective driving pressure (MSP – PRA) can be increased by reducing PRA (Fig. 3-1). The actual CO observed at any moment is defined by the intersection of Starling and venous return curves. Thus, both pump factors (heart rate [HR], loading conditions, and contractility) and circuit factors (intravascular volume, vessel tone) influence circulatory performance. Three basic mechanisms may cause or contribute to circulatory failure: (a) pump failure, (b) failure of vascular tone, and (c) hypovolemia.
Pump Failure
Heart Rate
CO, the product of heart rate (HR) and stroke volume (SV), can be depressed by abnormalities of either variable (see Chapter 1). To meet metabolic demand, isolated abnormalities of either SV or HR can be offset by adjustments in the other cofactor over a wide range. Both extremes of HR may cause CO to fall to shock levels. During sinus rhythm,
the maximal sustainable physiologic HR can be estimated as (HRmax = 220 − age). HRs that exceed this value may compromise CO and myocardial perfusion, even in healthy individuals. When the heart is noncompliant or compromised by coronary insufficiency, CO may fall at much lower HRs. Furthermore, the loss of atrial contraction that accompanies many tachyarrhythmias (i.e., atrial fibrillation) may depress CO on this basis alone.
the maximal sustainable physiologic HR can be estimated as (HRmax = 220 − age). HRs that exceed this value may compromise CO and myocardial perfusion, even in healthy individuals. When the heart is noncompliant or compromised by coronary insufficiency, CO may fall at much lower HRs. Furthermore, the loss of atrial contraction that accompanies many tachyarrhythmias (i.e., atrial fibrillation) may depress CO on this basis alone.
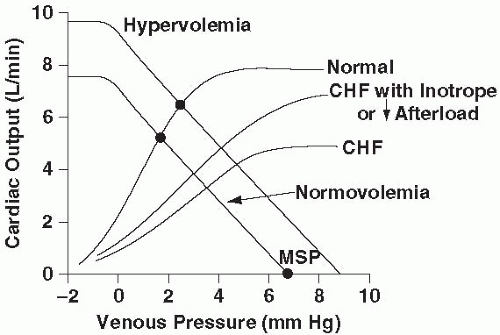 ▪ FIGURE 3-1 Regulation of CO. CO is determined by the intersection of the Frank-Starling and venous return curves. Venous return, which is driven by the difference between MSP and CVP, tends to improve as CVP falls, until the point at which venous pressure is insufficient to prevent vessel collapse (arrow). For the same venous return curve, the failing heart reduces its output, despite a higher filling pressure. CO can be maintained at a nearly normal level by increasing intravascular volume and/or using an inotrope or afterload reducer. |
In the intensive care unit (ICU), hypoxemia, enhanced vagal tone, and high-grade conduction block caused by intrinsic heart disease or pharmacologic agents are key mechanisms causing marked bradycardia. The normally compliant and contractile ventricle can adapt to physiologic or pathologic depressions in HR via the Starling mechanism; for example, young, well-conditioned athletes often maintain resting HRs less than 40 beats/min. However, patients with impaired myocardial contractility or effective compliance (e.g., ischemia, diastolic dysfunction, pericardial disease) may suffer marked depressions in CO and BP when HRs fall into the low normal range (<50 beats/min). This is especially true when the normally coordinated activation sequence is compromised or metabolic demands are high. Because bradycardia lengthens diastole as a proportion of total cycle time, MAP sinks closer to the diastolic value as HR slows. Perfusion of the heart and other key organs may suffer. It must be remembered that the appropriate physiologic response to hypovolemia is sinus tachycardia; hypotension suspected on the basis of massive gastrointestinal (GI) hemorrhage, therefore, should be accompanied by a compensatory tachycardia. A normal HR in this setting implicates an erroneous diagnosis or superimposed vagal, drug, or pathologic explanations.
Stroke Volume—Loading Properties and Contractility
SV is determined by end-diastolic volume and ejection fraction (see Chapter 1). End-diastolic volume, in turn, is the product of transmural filling pressure and myocardial compliance. As discussed in Chapter 1, SV and CO are often well preserved despite florid cardiogenic pulmonary edema. The myocardial hypertrophy of chronic hypertension or aortic stenosis, or the functional ischemia of coronary disease can result in symptomatic diastolic dysfunction. In such cases, control of hypertension and relief of ischemia and/or CO demand accelerate recovery and help prevent symptomatic recurrence.
Foremost among the primary depressants of contractility and ejection fraction during critical illness are (a) acute ischemia; (b) extensive myocardial necrosis; (c) humoral mediators (collectively known as “myocardial depressant factors”) released during inflammation and trauma; and (d) drugs that impair contractility (e.g., certain β-blockers, calcium channel blockers, and antiarrhythmics— most notably of the type Ia class). Cardiogenic shock consequent to acute myocardial infarction implies cumulative tissue losses exceeding 40% of the total myocardial mass.
Structural defects, such as papillary muscle rupture or post infarction ventriculoseptal defect (VSD), may compromise CO on a mechanical basis, even when there has been subcritical myocardial damage. Because the output of a failing heart is influenced by the impedance to ventricular ejection (afterload), increased vascular tone may improve BP at the expense of tissue perfusion. Patients with “tight” aortic stenosis are especially sensitive to changes in preload and contractility.
To scale for metabolic needs, the CO must be referenced to body surface area. The resulting quotient, the cardiac index (CI), attempts to take the mass of metabolizing tissue into account (normal >2.5 L /min /m2). CO adequacy can be judged only with respect to metabolic demands. Under some circumstances, even a normal CI may be insufficient for vital organ support. Such “high-output” cardiac failure can be precipitated by fever, anemia, thiamine deficiency, thyrotoxicosis, and arteriovenous shunting. Patients with extensive burns, severe sepsis, or cirrhosis also may have vastly higher CO requirements than the average resting patient.
Failure of Vascular Tone
Because organ perfusion depends on the gradient of pressure and the resistance to flow through the tissue bed, failure of vasomotor tone and/or distributive control may produce the shock syndrome, even when the CI is maintained within the normal range. Early sepsis provides a common example of maldistributive shock, characterized by reduced afterload and normal or elevated CO and [V with dot above]O2, despite undernourished vital tissue beds. General and spinal anesthesia, autonomic failure resulting from acute spinal cord injury, and certain drugs may also produce generalized, nonselective vasodilation that leads to underperfusion of critical organs. The inability to produce sufficient
glucocorticoid (cortisone), a common deficiency in critical illness (relative adrenal insufficiency), suppresses both vasomotor tone and myocardial contractility.
glucocorticoid (cortisone), a common deficiency in critical illness (relative adrenal insufficiency), suppresses both vasomotor tone and myocardial contractility.
The therapy of shock states often focuses on maintaining a targeted BP. This is an appropriate orientation, considering that the cerebral and coronary vasculatures (and to a lesser extent that of the kidney) are critically dependent on their perfusion gradients and relatively unaffected by the druginduced vasoconstriction experienced elsewhere. It must be emphasized that adequate BP and adequate flow are not synonymous; vasoactive drugs may cause intense ischemia of “nonvital” beds (gut, muscle, skin), occasionally with serious consequences for overall outcome.
Although moderate acidosis is generally well tolerated, severe metabolic acidosis may aggravate the shock state by causing myocardial depression, catecholamine resistance, increased right ventricular afterload, and potentially irreversible precapillary arteriolar dilation. Selective arteriolar dilation produces direct cellular injury and massive transudation of fluid into the extracellular space. Adrenal insufficiency and (less commonly) myxedema are two frequently overlooked endocrine problems that may contribute to vasomotor insufficiency and circulatory failure.
Hypovolemia
Although inadequacy of circulating blood volume is in itself a primary cause of circulatory failure, a relative deficiency of intravascular volume often contributes cause in the setting of impaired pump function or reduced vascular tone (e.g., sepsis). Primary hypovolemic shock develops during hemorrhage or when extensive extracellular volume losses result from burns, pancreatitis, vomiting, diarrhea, anaphylaxis, hypoproteinemia, or multiple traumas. Right ventricular infarction and pericardial disease mimic hypovolemia despite systemic venous congestion and/or normal wedge pressure because they impair left ventricular (LV) compliance.
Cardiodepression and Electrolyte Disturbances
Contractility of myocardium and vascular smooth muscle can be adversely influenced by nonphysiologic concentrations of key electrolytes, especially when several disorders are encountered simultaneously. Hypermagnesemia and hyponatremia occasionally are the primary causes of difficulty but usually serve a secondary role. The most frequent underlying causes of cardiovascular depression are hyperkalemia and deficiency of ionized calcium. An approach to the diagnosis and management of these disorders is provided in Chapter 13.
Pharmacologic Effects
Although all antihypertensive drugs may evoke hypotension, two commonly used classes of therapeutic agents—calcium channel blockers and β-blockers—directly interfere with cardiac function and may either exacerbate congestive heart failure (CHF) or encourage cardiovascular collapse. The appropriate response to excessive calcium channel blockade is to administer sufficient calcium to counter the drug’s adverse effect; either the chloride or gluconate salt can be given as a bolus. Calcium chloride can be administered by continuous infusion as well. Catechol-based vasopressors and inotropes are indicated in addressing hypotension, but all may prove ineffective until calcium channel blockade is overcome. High dose insulin (1-10 u/kg/hour) has shown promise in both emergency and experimental toxicology environments when glucose and potassium are kept within normal limits.
β Blockade
Through their actions on myocardial contractility, HR, bronchodilation, alveolar liquid clearance, and peripheral vasodilation, stimulation of one or more subtypes of β receptor is of fundamental importance to the recovery of the acutely compromised circulation. By influencing these properties, β-blocking drugs have unquestioned utility in managing a range of cardiovascular problems arising in the acute care setting, including ischemic cardiovascular disease, tachycardia, hyperthyroidism, aortic dissection, and acute hypertensive crisis. Whereas the use of well-selected β-blocking drugs is appropriate to the management of specific disorders that require such intervention, they should be withheld from the care of acutely ill patients in the absence of firm and specific indications (acute myocardial ischemia, symptomatic tachyarrhythmia, etc.). Currently, β-blocking drugs are frequently used in the outpatient setting not only for hypertension, but also to address diastolic dysfunction or chronic CHF (where they may help structural adaptation or remodeling). This practice, although partially justified by outcome studies of selective β-blockers in large populations, may backfire during periods of stress (e.g., sepsis, myocardial infarction, dietary indiscretion), especially in the elderly. The β-blocking drug already “on board” then accentuates
hypotension or contributes to “flash pulmonary edema” by interfering with the compensatory mechanisms of these patients with compromised cardiovascular reserve (Table 3-1). Certain long-acting, nonselective, and inexpensive β-blockers (e.g., atenolol) are especially problematic in that native drug and/or active metabolites are renally excreted and therefore accumulate during prerenal azotemia. The approach to managing life-threatening β receptor blockade is similar to that just described for calcium channel blockade: high dose insulin may be considered if β-stimulants, vasopressin and glucagon prove inadequate to reverse shock.
hypotension or contributes to “flash pulmonary edema” by interfering with the compensatory mechanisms of these patients with compromised cardiovascular reserve (Table 3-1). Certain long-acting, nonselective, and inexpensive β-blockers (e.g., atenolol) are especially problematic in that native drug and/or active metabolites are renally excreted and therefore accumulate during prerenal azotemia. The approach to managing life-threatening β receptor blockade is similar to that just described for calcium channel blockade: high dose insulin may be considered if β-stimulants, vasopressin and glucagon prove inadequate to reverse shock.
TABLE 3-1 POTENTIALLY ADVERSE ACTIONS OF β BLOCKADE | ||||
|---|---|---|---|---|
|
Effect of Shock on Organ Function
The closely autoregulated central nervous system of the healthy subject can tolerate marked reductions in MAP (to 50 to 60 mm Hg) without sustaining irreversible tissue damage. However, cerebrocortical functions are among the first to be impaired as shock develops.
As a rule, serious reductions of MAP are tolerated poorly by the GI tract. Early in shock, the gut suffers marked reductions in flow. This flow reduction eventually impairs mucosal function and bowel integrity, occasionally to the point of frank ischemic necrosis. A reduction of gastric mucosal pH is among the first indications of inadequate gut perfusion. One popular paradigm suggests translocation of bacteria across the abnormally permeable gut mucosa and into the lymphatic system or bloodstream as the next step on the path to multisystem organ failure. Hepatic ischemia may elevate liver function tests, alter the metabolism of drugs, and impair removal of toxins, lactate, and coagulation products.
Shock often impairs the clotting system sufficiently to initiate disseminated intravascular coagulation (DIC). The stimulus is multifactorial, and the important contributing factors are vascular endothelial injury, cell death, and impaired hepatic clearance of fibrin degradation products (see Chapter 30). In response to hypotension, the kidneys secrete renin to retain sodium and water. Intense vasoconstriction of the afferent arterioles shunts blood from the cortex to the medulla, reducing glomerular filtration to a greater degree than total renal blood flow or CO. If profound or prolonged, underperfusion may culminate in acute tubular necrosis. Antidiuretic hormone released from the pituitary helps conserve water and may contribute to hyponatremia.
During protracted shock, the skeletal muscles may release sufficient quantities of myoglobin into the circulation to impair renal tubular function (rhabdomyolysis). The hyperpnea that accompanies profound hypotension requires the respiratory muscles to consume large quantities of oxygen, outstripping the heart’s ability to deliver adequate flow to them, and ventilatory failure may result. Intubation and mechanical ventilation during circulatory shock decrease respiratory muscle O2 consumption, thereby increasing the blood flow available to other critical organs. Thus, in the vigorously breathing patient, mechanical ventilation often improves circulatory homeostasis.
Evaluation of Perfusion Adequacy
The history, physical examination, laboratory tests, and ancillary tests form the core of the evaluation process of the patient with circulatory inadequacy (Table 3-2). The clinician should rapidly undertake a targeted history, with review of prior vital signs and recent events and/or interventions that led to the shock state. Special attention is paid to preexisting illness and the medication listing.
Key features of the physical examination include otherwise unexplained alterations of vital organ function (mental status, urine output); hypotension relative to the usual baseline; sluggish capillary refilling; and cool, clammy skin. Neck vein examination and cardiac auscultation are essential focus points, keeping alert for signs of isolated right ventricular failure. Electrocardiogram (and in most cases echocardiogram) should be obtained in shock that is not of obvious cause or that does not reverse easily. Laboratory tests that reflect cardiac dysfunction (troponin, brain natriuretic peptide [BNP]) and/or perfusion inadequacy (anion gap, lactate) are often worth repeating as therapy progresses. Relative adrenal insufficiency is common enough to warrant measurement of serum cortisol concentration and its response to stimulation, especially in those refractory to intravenous fluid and vasopressor resuscitation. Arterial blood gas and central venous oxygen saturation data are sufficiently valuable that insertion of catheters should be considered to provide for their serial monitoring.
Key features of the physical examination include otherwise unexplained alterations of vital organ function (mental status, urine output); hypotension relative to the usual baseline; sluggish capillary refilling; and cool, clammy skin. Neck vein examination and cardiac auscultation are essential focus points, keeping alert for signs of isolated right ventricular failure. Electrocardiogram (and in most cases echocardiogram) should be obtained in shock that is not of obvious cause or that does not reverse easily. Laboratory tests that reflect cardiac dysfunction (troponin, brain natriuretic peptide [BNP]) and/or perfusion inadequacy (anion gap, lactate) are often worth repeating as therapy progresses. Relative adrenal insufficiency is common enough to warrant measurement of serum cortisol concentration and its response to stimulation, especially in those refractory to intravenous fluid and vasopressor resuscitation. Arterial blood gas and central venous oxygen saturation data are sufficiently valuable that insertion of catheters should be considered to provide for their serial monitoring.
TABLE 3-2 VITAL DATABASE IN HYPOTENSIVE STATES | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
▪ THERAPY OF THE FAILING CIRCULATION
Goal-Directed Therapy
Because prolonged hypoperfusion leads to sustained organ failure and irreversibility, most experienced clinicians agree that shock should be reversed as quickly as feasible to safely do so. Early aggressive intervention to restore an effective circulation has become an accepted goal, even if the precise target at which to aim is debated. Two elements seem important: early intervention and restoration of adequate perfusion to vital organs. What measurable indicator to shoot for? Generally speaking, supranormal values for CO and oxygen delivery are neither easy to achieve nor needed. Restoring near-normal values for central venous O2 saturation, however, currently is thought prudent, based on the persuasive results of an important prospective clinical trial. The central venous saturation reflects the ratio of O2 delivery to consumption. Targeting a central venous saturation of >70% has been associated with higher survival than use of an MAP target. Unlike the mixed venous O2 saturation, central venous sampling can be readily achieved via routine central lines as well as monitored continuously via specialized catheters. As a helpful but not infallible indicator of perfusion adequacy, central venous O2 saturation should be considered a valuable complement to routine arterial pressure monitoring.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


