Ethanol ingestion may cause acute gastritis and gastrointestinal (GI) bleeding. Alcoholics have an increased incidence of peptic ulcer disease and pancreatitis; acute alcohol intoxication may precipitate alcoholic hepatitis in the chronic user. All bone marrow cell lines are suppressed by alcohol ingestion, and suppression of antidiuretic hormone by ethanol causes diuresis and may lead to profound hypovolemia, especially if there is associated nausea, vomiting, or diarrhea.
Assessment and Treatment of Acute Intoxication
Treatment of acute ethanol intoxication is primarily supportive, but a careful examination is needed to detect complications or concomitant conditions. The first priority is assessment and stabilization of the airway and ventilation. The respiratory rate, depth of respirations, SpO2, mental status, and gag reflex should be rapidly evaluated, as should evaluation for the presence of vomitus. Arterial blood gas analysis should be obtained if hypoventilation is a concern, but is not obvious on clinical examination. Endotracheal intubation is indicated in the obtunded or comatose patient unable to protect his/her airway, or when aspiration has occurred or is likely. Positive pressure ventilation should be instituted to correct alveolar hypoventilation and hypoxemia. If the patient presents with altered mental status, 50 to 100 mg of thiamine and 25 g of glucose should be administered intravenously. Thiamine should be administered before glucose to avoid precipitation of acute beriberi and Wernicke–Korsakoff syndrome. If the patient responds to the administration of glucose or if blood glucose levels are low, a continuous infusion of glucose should be given. Intravenous (IV) naloxone may be administered if concomitant opioid use is suspected. Hypotension should be treated initially with volume resuscitation. GI bleeding should be considered in the hypotensive patient and further assessment may include a rectal examination and insertion of a nasogastric tube. GI decontamination is of limited utility because the majority of alcohol is already absorbed; ethanol is not adsorbed by activated charcoal, but charcoal may be administered if ingestion of other toxic drugs is suspected. Hypothermia should be corrected, as should be fluid, electrolyte, and acid–base disturbances, depending on the clinical presentation. A creatine phosphokinase (CPK) level may be warranted in the patient with trauma or prolonged muscle compression to evaluate for rhabdomyolysis. An ethanol blood level may be helpful in documenting the severity of intoxication and estimating the duration of impairment. The serum osmolar gap may be evaluated with an adjustment for blood ethanol level if coingestion of other alcohols is suspected. A low ethanol level in the setting of a patient with a depressed level of consciousness should prompt an evaluation for other etiologies. A chest radiograph is often necessary to assess for evidence of aspiration or other complications, such as pneumonia. Consider obtaining CT of the head if there is any suspicion of subdural hematoma or other intracranial injury. Pancreatitis should be promptly treated with IV hydration and supportive care.
Alcohol Withdrawal
Chronic excessive alcohol ingestion depresses central α- and β-receptors and potentiates the inhibitory neurotransmitter γ-aminobutyric acid (GABA). The brain adapts with a functional increase in n-methyl D-aspartate (NMDA) receptors, which are part of an excitatory system. When alcohol consumption stops, the excess excitatory receptors and removal of the inhibitory effects mediated by GABA contribute to the hyperadrenergic state that causes the symptoms seen in alcohol withdrawal.
Alcohol withdrawal syndromes occur in dependent patients during the initial period of abstinence. The incidence of alcohol withdrawal syndromes in hospitalized and critically ill patients is difficult to assess due to varying definitions, patient populations, and type of ICU (6). Prevention of alcohol withdrawal syndromes has been shown to improve morbidity and mortality and to shorten hospital and ICU length of stay (7). Four stages of alcohol withdrawal have been described (8), but symptoms are a continuum of neuropsychiatric and hemodynamic manifestations. Patients may manifest one or more of these syndromes on presentation or develop additional manifestations and progress from less severe to more severe stages while hospitalized (Table 152.2). A key distinction is to determine if the patient has an intact or altered sensorium.
Assessment of the severity of withdrawal is needed to determine appropriate treatment. Although the revised Clinical Institute Withdrawal Assessment-Alcohol Scale (CIWA-Ar) is often used for assessment, it has limited applicability in critically ill patients and has not been validated in this patient population (9). Appropriate use of the CIWA-Ar requires recent alcohol use and the ability to communicate and these requirements result in misuse in many hospitalized patients (10).
| TABLE 152.2 Stages of Alcohol Withdrawal and Treatment | ||
 | ||
Patients with minor withdrawal symptoms—tremulousness and hallucinosis—can usually be treated with IV or oral benzodiazepines. Benzodiazepines act as an alcohol substitute to dampen the excitatory neuronal activity, and additional benefits include prevention of seizures and delirium tremens (DTs). The choice of benzodiazepine in hospitalized patients may depend on severity of hepatic dysfunction, desired duration of action, and available routes of administration. While all benzodiazepines are effective when appropriate doses are used, high doses may cause respiratory depression. Fixed dosing and symptom-triggered regimens have been used effectively. Fixed dosing may be more appropriate in critically ill patients until other conditions have stabilized; treatment duration beyond 7 days is seldom required. Frequent reassessment and intervention is essential in the first 24 to 48 hours of withdrawal symptoms to prevent progression to DTs.
Benzodiazepines are clearly superior to placebo in treating alcohol withdrawal and also superior for seizure control when compared to baclofen, γ-hydroxybutyrate, anticonvulsants, and the psychotropic analgesic nitrous oxide (11). Many trials have been conducted in outpatients and have limited applicability to hospitalized and critically ill patients. IV ethanol may also be an option for alcohol withdrawal treatment or prophylaxis (12). However, it is not recommended for routine use due to dosing variability and lack of established efficacy (13). Other agents such as clonidine and β-blockers have been reported to be effective for minor withdrawal symptoms, but their use is less common; they do not prevent the development of delirium. All patients with alcohol withdrawal should receive supportive measures in addition to pharmacologic intervention. Thiamine (vitamin B1) should be given intravenously or orally to prevent Wernicke encephalopathy. Magnesium sulfate may be needed to correct hypomagnesemia but there is no evidence that it prevents or treats alcohol withdrawal (14).
Seizures
Approximately 5% to 10% of patients with untreated mild alcohol withdrawal symptoms progress to seizures, but seizures frequently occur in the absence of other withdrawal symptoms. Patients who have been drinking heavily for only a few years, but have several detoxification admissions are at higher risk of seizures than patients with long drinking histories but fewer detoxification admissions. Previous nonalcohol-related admissions also increase the risk of alcohol withdrawal seizures. This association has been termed the “kindling effect.” According to the kindling hypothesis, each withdrawal episode is an irritative phenomenon to the brain. The accumulation of multiple episodes lowers the seizure threshold (15). Most alcohol withdrawal seizures are brief and self-limited in duration. Alcohol withdrawal seizures are usually generalized tonic–clonic, but focal seizures may also occur. Multiple seizures—two to six episodes—occur in approximately 60% of patients and within a 12-hour period. It may be difficult to distinguish withdrawal seizures from a pre-existing seizure disorder or new onset of a nonalcohol-related seizure. Other causes of seizures such as hypoglycemia, metabolic abnormalities, trauma, infection, and other drug intoxication must be considered. A computed tomography (CT) scan of the head should be obtained for new-onset seizure, persistent neurologic deficits, or evidence or suspicion of trauma. Treatment is aimed at terminating active seizures and preventing subsequent seizures. Benzodiazepines have been shown to prevent withdrawal seizures compared to placebo and are used to terminate seizures (16). IV lorazepam or midazolam is commonly used; as the risk of a recurrent seizure is 13% to 24%, additional prophylactic treatment is indicated (17). Lorazepam (2 mg) significantly reduces the risk of recurrent seizure, whereas phenytoin has no effect (18). Less than 3% of patients develop status epilepticus and they should be treated with benzodiazepines or propofol. Phenytoin is not as effective.
Delirium Tremens
DTs is the most severe manifestation of alcohol withdrawal and these patients should be cared for in an ICU setting. Untreated DTs carries a mortality of 15%, declining to 1% if treated. The accumulation of multiple prior withdrawal episodes leads to more severe DTs with each episode (15). Patients with DTs have more severe autonomic hyperactivity than milder stages of withdrawal and manifest delirium that may fluctuate. Some patients with severe withdrawal symptoms may need intubation during treatment. Fluid requirements may be increased due to increased insensible losses (fever, diaphoresis) and lack of oral intake. High-dose IV benzodiazepines (diazepam, lorazepam, midazolam) administered at frequent intervals or as a continuous infusion are needed to control the hyperadrenergic symptoms. Dosing should be individualized to achieve light somnolence (19,20). Benzodiazepines bind at the GABA–benzodiazepine receptor, and once these receptors are saturated, additional drug cannot bind. Thus, patients may tolerate high doses of benzodiazepines but do not necessarily benefit from them (21). Caution is advised when administering high doses of IV lorazepam or diazepam over long periods of time as the propylene glycol diluent may result in a lactic acidosis (22). Daily dose reductions of 25% can be initiated after the second or third day of treatment.
Dexmedetomidine, a central α2-adrenergic agonist, and propofol have been used as adjunctive therapy with benzodiazepines in severe withdrawal in critically ill patients (23–26). One prospective study evaluating dexmedetomidine found decreased use of benzodiazepines in the short term but not long term, no difference in intubation or seizures, and a higher incidence of bradycardia (27). Propofol infusions may be useful for patients who are refractory to benzodiazepines as the former has a dual activity similar to alcohol (GABA agonist and NMDA antagonist properties) that may explain its efficacy. Propofol has a rapid onset of action, sedation, and anticonvulsive properties, but its use requires intubation and mechanical ventilation (28). Other adverse effects of propofol include hypotension, bradycardia, and propofol infusion syndrome. Other sedative–hypnotic drugs such as paraldehyde and barbiturates are effective in treating DTs but are not commonly used. Neuroleptic agents are inferior to benzodiazepines and should not be used as single agents for treatment of DTs (20). Neuromuscular blockers may be considered to control agitation when high-dose sedatives are not effective. Cardiac monitoring is necessary to detect dysrhythmias early and institute therapy. Torsade de pointes may develop due to hypomagnesemia and/or prolongation of the QTc interval and should be treated aggressively with IV magnesium sulfate. β-Blockers may be needed to treat hypertension or tachycardia but they should not be administered to treat delirium; propranolol may worsen delirium. Thiamine supplementation (100 mg/d) is recommended for 3 days.
DTs usually lasts 2 to 5 days, but in 5% to 10% of cases, DTs lasts greater than a week. Elderly alcoholics have a longer withdrawal period with more symptoms than do younger ones (29). A small percentage of patients remain delirious for several weeks and require continuing treatment. Be aware, however, that after head trauma, a subdural hematoma can evolve subacutely in the alcoholic patient; repeat imaging of the brain may be warranted 7 to 10 days into a course of protracted delirium to rule out a slowly accumulating subdural hematoma (21).
COCAINE
Cocaine is the third most commonly used illicit drug after marijuana and prescription-type drugs (psychotherapeutics) (1). Cocaine (benzoylmethylecgonine) is an alkaloid derived from leaves of Erythroxylon coca and it is available in two forms. Cocaine hydrochloride is prepared by dissolving alkaloidal cocaine in hydrochloric acid resulting in a white water-soluble powder, crystals, or granules. This form of cocaine is used intranasally (snorting), orally, or intravenously. The other available form of cocaine is free base or crack cocaine. Heating cocaine hydrochloride in sodium bicarbonate or ammonia makes the hard crystallized cocaine base called crack because of the popping sound it makes when heated. Smoking crack cocaine has become a widespread practice due to the rapid absorption across the alveolar surface. Both forms of cocaine are readily absorbed from all body mucosal surfaces. The peak effects of cocaine range from 1 to 90 minutes depending on the route of administration. Inhalational and IV use result in the most rapid peak effects and the shortest duration of action. Cocaine is rapidly metabolized by hepatic and plasma cholinesterases and nonenzymatic hydrolysis to ecgonine methylester and benzoylecgonine, which are excreted in urine; the urinary excretion of unchanged cocaine ranges from 1% to 15%. The route of administration does not affect metabolic excretion patterns appreciably and half-lives of most metabolites range from 45 to 90 minutes (30). Subjective rating of euphoria declines within minutes after constant concentrations are achieved, demonstrating rapid desensitization and acute tolerance (31). Duration of positive urinary metabolites is somewhat dependent on the assay technique, the activity of plasma cholinesterases, and the duration and dosing of cocaine use.
Cocaine’s lipophilic nature, compounded with rapid distribution into and out of the CNS, suggests a highly abusive profile (rush and crash) and increased incidence of kindling. The major neurochemical actions of cocaine are CNS stimulation with release of dopamine; inhibition of neuronal norepinephrine and dopamine uptake, resulting in generalized sympathetic nervous system stimulation; release of serotonin or blockade of serotonin reuptake; and inhibition of sodium current in neuronal tissue, resulting in a local anesthetic effect.
Toxicity
Numerous morbidities have been associated with acute and chronic cocaine use (Table 152.3). Complications of particular interest to intensivists are discussed below (32).
| TABLE 152.3 Clinical Manifestations of Cocaine Use |
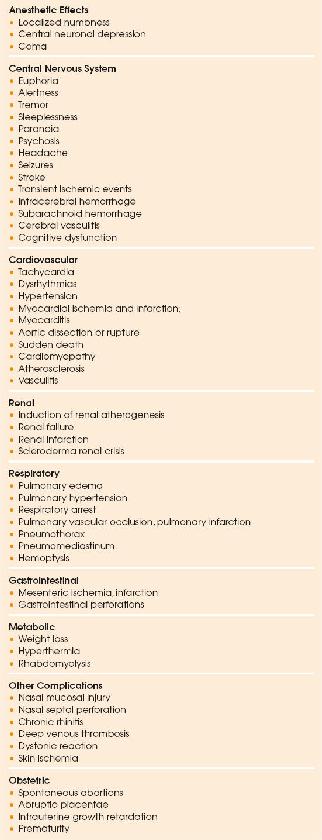 |
Cardiovascular
Cocaine increases the heart rate, blood pressure, and left ventricular contractility, leading to an increase in myocardial oxygen demand (33). The increased demand may combine with underlying coronary artery disease, vasoconstriction, platelet aggregation, or in situ thrombus formation to produce ischemia and infarction. Chronic cocaine use also accelerates atherosclerosis (34). Apart from structural changes in epicardial vessels, wall thickening is described in the intramyocardial small coronary arteries in people with cocaine-induced chest pain (35).
Chest pain is the most common cocaine-associated complication in patients who present for medical care. All patients presenting with chest pain should be questioned regarding cocaine use. Myocardial ischemia can occur with all routes of abuse with no relation to the dose or chronicity of use. The onset of chest pain often occurs temporally related to the use of cocaine. However, chest pain may occur hours to days after the last use of cocaine. Electrocardiograms (ECGs) are often abnormal in patients presenting with cocaine-associated chest pain (36,37). Myocardial infarction may be present with a normal or abnormal ECG. Conversely, ECGs may suggest acute ischemia in the absence of infarction due to J-point elevation or repolarization changes (37). Cardiac troponins are more specific for assessing myocardial injury than creatine kinase-MB, which may be elevated due to skeletal muscle injury (38). Myocardial infarction is reported to occur in approximately 6% to 7% of patients and occurs with normal coronary arteries and in the presence of significant atherosclerotic disease (37,39,40). Periods of silent ischemia are common in chronic users of cocaine, as shown by ambulatory monitoring and during periods of withdrawal (41). Dilated cardiomyopathy, myocarditis, and congestive heart failure can occur secondary to chronic cocaine use (42).
Cocaine is dysrhythmogenic when taken in large quantities because of catecholamine effects. The dysrhythmias are usually transient and resolve when cocaine is metabolized. Sinus tachycardia, supraventricular tachycardia, atrial fibrillation, premature ventricular beats, ventricular tachycardia, ventricular fibrillation, bundle branch block, asystole, and torsade de pointes may occur.
Elevation of blood pressure occurs due to the acute effects of cocaine, but it is usually self-limited. Sustained elevations of blood pressure suggest the presence of chronic hypertension or another complication (e.g., intracranial process). The elevations of blood pressure may contribute to other catastrophic complications such as stroke and intracranial hemorrhage. Rupture of the ascending aorta in previously healthy individuals has been reported as well as aortic dissection (43).
Central Nervous System
In large doses, cocaine may cause a generalized impairment of neuronal impulse transmission leading to CNS depression, coma, respiratory depression, and respiratory arrest. At low doses, stimulation is the common feature of cocaine use. The euphoria produced by cocaine is the principal reason for its abuse. Excessive CNS stimulation can occur and is manifested by tremulousness, agitation, sleeplessness, paranoia, and frank psychosis; aggressive and assaultive behavior can occur in cocaine overdose.
Seizures can be induced, even on the first exposure, because cocaine lowers the threshold for seizures. Cocaine-related seizures are usually brief and self-limited, occurring soon after taking cocaine, although the interval between the last use of cocaine and the onset of seizures can be several hours (44). Sustained or repeated seizure activity suggests an additional complication such as hyperthermia, intracranial hemorrhage, metabolic abnormality, or massive intake of cocaine.
Cocaine use is associated with ischemic cerebrovascular accidents as well as transient ischemic attacks (44–46). Radiologic studies have demonstrated cerebral vasoconstriction as well as vessel thrombosis with cocaine (46,47). Although most symptoms occur during or immediately after cocaine use, neurologic symptoms may occur within hours to several days after the last use. Subarachnoid, parenchymal, and intraventricular hemorrhage may occur within moments of drug use, possibly related to blood pressure elevation. Some patients have anatomic abnormalities such as vascular malformation or aneurysm that may be amenable to specific therapy (44,48,49). Cerebral atrophy, predominantly in the temporofrontal regions, has been noted in patients with chronic cocaine abuse (50).
Pulmonary
Pulmonary complications associated with cocaine are much less common than cardiovascular and cerebrovascular events but include a variety of conditions (51,52). Inhalation of cocaine, in contrast to IV use, has been demonstrated to cause bronchoconstriction (53). This response may be due to an irritant effect and may contribute to wheezing and exacerbations of asthma in cocaine users (54,55). Barotrauma (pneumothorax and pneumomediastinum) is reported secondary to snorting cocaine and crack inhalation (56). Noncardiogenic pulmonary edema may occur and is described more commonly with the IV use of cocaine. Massive hemoptysis with diffuse alveolar hemorrhage is a rare complication of unknown etiology and has been reported with smoking free-base cocaine and other routes of abuse. Other rare pulmonary toxicities, more commonly reported after inhalation of cocaine, include interstitial pneumonitis, pulmonary infiltrates with peripheral and/or lung eosinophil prominence, and bronchiolitis obliterans (57). Septic pulmonary emboli and pulmonary vascular obstruction resulting from foreign body granulomas or angiothrombosis may develop as a consequence of IV cocaine use similar to IV heroin use (58).
Hyperthermia/Rhabdomyolysis
Hyperthermia may result from muscle hyperactivity or as a direct effect of cocaine on the hypothalamic temperature regulatory center. High ambient temperatures are associated with increased mortality from cocaine and hyperthermia is probably one of several factors that play a role (59). Cocaine impairs sweating and cutaneous vasodilation as well as heat perception under conditions of heat stress (60).
Cocaine-induced rhabdomyolysis is common and can lead to acute renal failure. Multiple factors such as hyperthermia, seizures, vasoconstriction with ischemia, excessive motor activity, concomitant use of other drugs, and even a direct toxic effect of cocaine may contribute to muscle injury; myalgias and muscle tenderness are infrequently present. Seizures, hypotension or hypertension, dysrhythmia, coma, and cardiac arrest identify a subgroup of patients who are prone to severe rhabdomyolysis (61,62).
Other Toxicities
Intestinal ischemia, infarction, and perforation have been reported following ingested, IV, and inhaled cocaine (63,64). Patients may present with complaints of acute or chronic abdominal pain. Acute renal failure may be precipitated by rhabdomyolysis, but other etiologies may include accelerated hypertension and glomerulonephritis (65). Rare cases of renal infarction have also been reported.
Coingestions and Adulterants
Cocaine may be abused with other drugs such as marijuana, opioids, amphetamines, ethanol, and benzodiazepines, and the toxicities of these drugs may also be evident. Cocaine and ethanol when consumed together form a more toxic compound, cocaethylene, in the liver; it produces more euphoria and has a longer duration of action (66).
There are several substances such as talc, benzocaine, phenacetin, and thallium that may be used as adulterants of cocaine. Levamisole is a more recent adulterant found in the cocaine supply (67). Levamisole is an immunomodulatory agent and commonly used as an anti-helminth drug. It can cause agranulocytosis, arthralgias, vascultitis, retiform purpura, and skin necrosis (especially on ear lobes and the nasal tip). Levamisole can be detected using gas chromatography/mass spectrometry.
Diagnosis of Acute Intoxication
Patients with cocaine intoxication may present with a variety of primary complaints such as altered mental status, chest pain, syncope, palpitations, seizures, or attempted suicide. Characteristic findings of CNS stimulation such as agitation, mydriasis, sweating, hypertension, and tachycardia are often present. However, the effects of other drugs, the presence of complications, and delays in presentation may obscure the typical sympathomimetic manifestations. Other medical conditions such as meningitis, encephalopathy, epilepsy with status, and thyrotoxicosis may mimic cocaine intoxication (68). Confirmation of acute or recent cocaine exposure is made by urine toxicology testing.
Treatment for Acute Intoxication
Benzodiazepines are the pharmacologic agents of choice for control of cocaine-induced agitation. The agitation and psychosis of cocaine overdose usually can be managed with titrated doses of IV diazepam, 5 to 20 mg; lorazepam, 2 to 4 mg; or midazolam, 5 to 10 mg slowly. Haloperidol is not recommended as a first-line agent because of the lack of experimental support (69) and its potential to lower the seizure threshold. Although low-dose dexmedetomidine may decrease some sympathetic findings in an experimental setting of inhaled cocaine use, it has not been evaluated in acute intoxications (70,71). Adequate hydration and correction of electrolyte abnormalities are important.
Cardiovascular
No large clinical trials have evaluated treatment strategies for cocaine-associated ischemia. Treatment of cardiac toxicity due to cocaine is directed at reversing physiologic effects that cause ischemia or dysrhythmias. Aspirin should be administered as an antiplatelet agent for suspected myocardial ischemia unless there is evidence of cerebral hemorrhage. Oxygen may also help to limit myocardial ischemia. Benzodiazepines and nitroglycerin are considered first-line agents for relief of chest pain, but small clinical studies have yielded conflicting results on the benefit of combining the agents (72,73). Benzodiazepines decrease the blood pressure and heart rate, thus decreasing myocardial oxygen demand, and nitroglycerin may dilate coronary arteries or relieve vasoconstriction. α-Blockers such as phentolamine have been recommended as a second-line treatment for unrelieved pain, but are rarely needed (74). The use of β-blockers in the management of myocardial ischemia is debated. There is a potential concern of worsening vasospasm or hypertension due to unopposed stimulation of α-receptors. Intracoronary propranolol results in a small decrease in coronary artery diameter following intranasal cocaine, but β-blockers are not administered by this route or as soon after cocaine use in most patients (75). However, β-blockers have been used, particularly in the setting of myocardial infarction, without complications. Administration of β-blockers might be avoided in patients manifesting acute sympathomimetic findings, but the benefits of these agents should be considered in other patients with ongoing myocardial ischemia.
Most patients with cocaine-associated chest pain will not have infarction. Patients can be managed in chest pain or observation units similar to other chest pain patients (76). Low-risk patients with normal cardiac markers can be risk stratified safely with stress testing.
Early therapy for cocaine-induced myocardial infarction should consist of oxygen, aspirin, and nitroglycerin as required for pain relief (77). The role of calcium channel blockers remains uncertain but they should not be used as first-line therapy. β-Blockers should be avoided in the presence of acute sympathomimetic symptoms. If pain persists, patients with cocaine-induced myocardial infarction are candidates for reperfusion therapy. Percutaneous coronary intervention (PCI) is preferred in patients with evidence of ST-elevation myocardial infarction, especially when the diagnosis may be in doubt (77). Thrombolytic therapy has been safely used in cocaine-associated myocardial infarction but should be reserved for patients who cannot receive PCI due to the risk of intracranial hemorrhage (78).
Dysrhythmias associated with cocaine use are usually transient. Standard therapy should be considered for sustained dysrhythmias unresponsive to control of pain and agitation. Although lidocaine is seldom used for ventricular dysrhythmias, theoretical concerns of enhancing cocaine toxicity do not appear to be clinically significant (79).
Sustained hypertension in acute cocaine intoxication is not common due to the short physiologic effects of the drug. Control of agitation with benzodiazepines often results in resolution of hypertension. IV labetalol is a reasonable option if the blood pressure needs to be lowered due to its combined α- and β-blocking effects; nicardipine and nitroglycerin are also reasonable options for lowering blood pressure. Cocaine-intoxicated patients should be considered to have acute elevations in blood pressure, and unless there is documentation or clinical evidence of long-standing hypertension, there should be little concern about cerebral hypoperfusion with immediate lowering of blood pressure to normal levels (80).
Central Nervous System
Seizures induced by cocaine are best controlled with IV benzodiazepines. Other standard antiepileptics can be added for refractory cases. If neuromuscular blockers are used, seizure activity may persist yet be unrecognized; hence, the use of this drug class warrants continuous electroencephalographic monitoring.
Interventions for ischemic strokes associated with cocaine use should be carefully considered. Since the etiology may involve vasoconstriction as well as thrombosis, the decision to use thrombolytic agents in patients presenting within 3 hours of symptom onset may be more difficult. Vascular imaging, if readily available, may be helpful. Blood pressure is not usually severely elevated, but if sustained hypertension is present, current guidelines should be followed for lowering blood pressure. Neurosurgical consultation should be sought for intracranial hemorrhages to evaluate for possible interventions. Patients with subarachnoid hemorrhage should be evaluated for vascular malformations that may be amenable to treatment.
Pulmonary
Most pulmonary toxicities associated with cocaine are managed with usual care or supportive care (51,52). Bronchospasm and asthma should be treated with inhaled β-agonists and corticosteroids, if indicated. Pneumomediastinum can be followed without hospital admission for most patients. Small pneumothoraces may also resolve without intervention, whereas large pneumothoraces will require tube thoracostomy. Noncardiogenic pulmonary edema may require supplemental oxygen and mechanical ventilation, but resolves within a few days unless other complications occur.
Hyperthermia/Rhabdomyolysis
Hyperthermia associated with cocaine use should be treated aggressively by rapid cooling (see Chapter 68, Temperature Related Injuries). Control of coexisting agitation, psychosis, or seizures is essential to achieve and maintain cooling while avoiding brain, hepatic, and muscle cell destruction. There is no evidence that pharmacologic agents such as dantrolene are of benefit in cooling patients with life-threatening hyperthermia.
Patients with hyperthermia, severe agitation or motor activity, seizures, and obtundation should be evaluated for rhabdomyolysis. Aggressive fluid resuscitation to replete the intravascular volume and enhance urine output should often be initiated prior to definitive diagnosis. Serial tests of electrolytes, renal function, and CPK are needed to monitor the severity and response of rhabdomyolysis.
Body Packers/Stuffers
Individuals may ingest packets of cocaine or any illicit drug for the purpose of transport or concealment. Body stuffers swallow small amounts of drug (wrapped or unwrapped) in order to avoid arrest. In this circumstance, drugs are not prepared for passage through the GI tract and drug is frequently absorbed. Due to the smaller quantities of drug, toxicity is usually mild (81). In contrast, body packers swallow larger quantities of drug in multiple packets that are specially prepared for smuggling to withstand transit through the GI tract. Abdominal radiographs often show the location of the packets and allow tracking as they move through the GI tract. However, a negative result on plain abdominal radiograph does not rule out body packing, and an abdominal CT scan may be needed to visualize the packets (81).
Most body packers are asymptomatic and can be managed conservatively until the packets have been completely evacuated (81,82). Whole bowel irrigation may assist with passage of the packets. Body packers with signs and symptoms of drug toxicity, in vivo degradation, or GI obstruction require emergent surgical intervention (81).
Cocaine Withdrawal
Psychological and biochemical dependency on cocaine may be intense. Cocaine causes activation of the dopamine system and blocks dopamine uptake, especially in the pleasure centers of the brain (83). The brain becomes dopamine deficient, and even a short period of cocaine abstinence can result in a withdrawal state.
The clinical effects of cocaine withdrawal include depression, fatigue, irritability, sleep and appetite dysfunction, psychomotor agitation or retardation, and craving for more cocaine (30). A period of prolonged somnolence and decreased arousal can occur after the binge use of cocaine and often necessitates evaluations to rule out complications associated with cocaine use (84). A supportive environment and professional drug counseling are warranted.
OPIOIDS
Opioids include all drugs (synthetic as well as natural) that have morphine-like properties and/or bind to opioid receptors. There are at least five opioid receptors with various physiologic roles including analgesia, ventilatory depression, drug dependence, bradycardia, dysphoria, hallucinations, sedation, and miosis. Opioids are classified as receptor agonists or antagonists. Some have combined properties because they stimulate one type of receptor and antagonize another. A classification of opioids is found in Table 152.4. Opioid dependence is characterized by repeated self-administration of drug and encompasses physiologic dependence and addictive behavior. Exposure to opioids causes neural changes that produce tolerance, dependence, and withdrawal (85).
Toxicity
Abuse of prescription opioid pain relievers has been increasing, resulting in overdoses and fatalities (86,87). Heroin is rapidly absorbed by all routes of administration, including IV, intranasal, intramuscular, subcutaneous (skin popping), transdermal (fentanyl patches), and inhalation. Most fatal overdoses occur with IV administration; IV fentanyl extracted from analgesic patches is also associated with fatalities (88). Oral opioids are available illicitly or by prescription, and toxicity depends on the potency of the agent, dose ingested, and tolerance of the individual. Codeine elixir (“syrup”) is abused by adolescents and young adults. The diagnosis of opioid toxicity is made by characteristic clinical findings, exposure history, qualitative toxicology assay, and response to naloxone. Qualitative urine assays may not detect all opioid derivatives (e.g., fentanyl) and have little impact on immediate evaluation and treatment.
| TABLE 152.4 Classification of Opioid Agents |
 |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


