Country
Age-adjusted mortality rate per 100,000 per year
Canada
25.7
USA
30.4
Australia
32.6
United Kingdom
45.6
Kyrgyzstan
249.4
India
108
China
156.5
Russian Federation
228
South Africa
141.1
Nigeria
152.8
Brazil
91.1
Pakistan
117.9
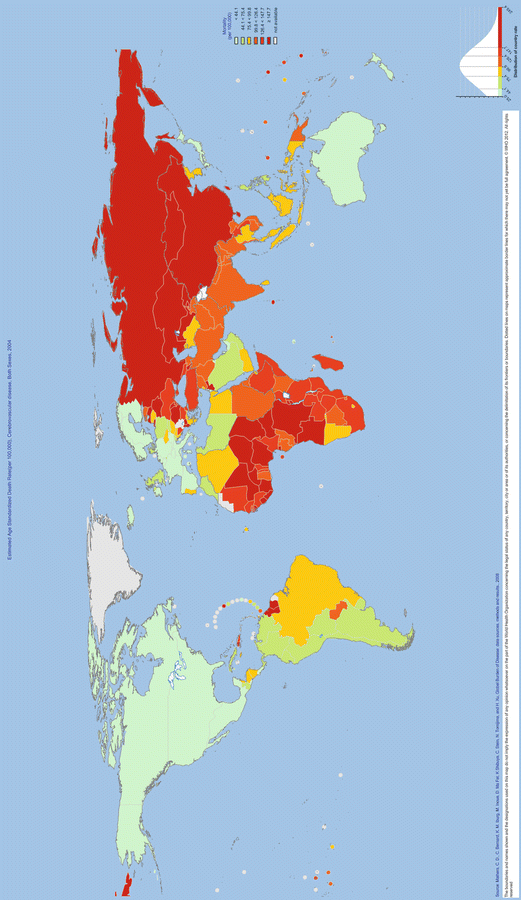
Fig. 1.1
WHO Global Infobase. Estimated age standardized death rate, cerebrovascular disease, both sexes, 2004. Reprinted with permission from the World Health Organization
Estimates suggest that stroke consumes 2–4 % of the total healthcare costs globally. In developed nations this proportion exceeds 4 % of healthcare costs [10]. In the United States, the estimated cost of stroke in 2008 was $34.3 billion; this includes direct medical costs of $18.8 billion [11].
United States data estimates the annual stroke incidence to be 795,000, of which approximately 610,000 are first-time strokes [12]. In the United States, stroke as a cause of death declined by 19 % from 1998 to 2008 so that it is now the fourth most common cause of death. The lifetime risk of stroke is greater for women than for men, and women are generally older at stroke onset. Racial differences have also been observed with African- and Mexican-Americans having a higher incidence of stroke than Caucasian-Americans [12].
Whilst cardio-embolic stroke and ischemic stroke incidence rates in general have been relatively stable or declining over the past few decades, the incidence of intracerebral hemorrhage has not changed and anticoagulant-associated intracerebral hemorrhage has been increasing. This trend will likely continue in developed countries as the average age of the population increases and there is increasing use of anticoagulants. Whether new oral direct thrombin inhibitors such as dabigatran and oral factor Xa inhibitors such as apixaban and rivaroxaban, which may be associated with lower rates of intracranial hemorrhage, will prevent this is unclear [13].
Etiology
Stroke—by definition a loss of brain function due to disturbance in blood supply to the brain—has been classified based on the duration of symptoms, clinical presentation, etiology, vascular or anatomical area of the brain affected, and by syndrome. About 80–85 % of strokes are ischemic and 15–20 % are hemorrhagic. The causative mechanism may be related to local intracranial vascular pathologies such as atherosclerosis, a more distant source of pathology such as emboli from extracranial artery disease or the heart, or reduced cerebral perfusion in the setting of circulatory failure. These categories are further subdivided based on the causative process (Fig. 1.2).
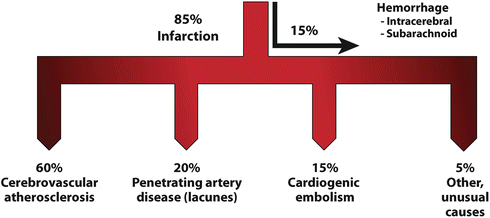
Fig. 1.2
Stroke etiologies
Ischemic stroke may be caused by large artery thrombosis, cardio-embolism, small vessel occlusion, venous infarction, or other determined or undetermined causes. Hemorrhagic stroke may be caused by hypertension, vascular malformations, amyloid angiopathy, an infarct with secondary hemorrhage, spontaneous subarachnoid hemorrhage usually due to aneurysm rupture, anticoagulants or antiplatelet drugs, coagulation and platelet disorders, and a small proportion of them are of undetermined etiologies.
Large vessel disease encompasses pathology involving the extracranial carotid vessels and the intracranial arteries including the proximal branches of the Circle of Willis. Pathological processes affecting large arteries include atherosclerosis, arterial dissection, inflammatory arteritis, vasculitis, fibromuscular dysplasia, Moyamoya disease, and cerebral angiographic vasospasm after subarachnoid hemorrhage. Atherosclerosis is the predominant cause of large vessel disease. Thrombi can form on atherosclerotic plaques or spontaneously in the setting of an underlying coagulopathy. Identification of the specific underlying vascular pathology and its extent is integral to determining the potential value of subsequent surgical and medical interventions.
Transient ischemic attacks (TIAs) are “a falsely benign form of brain attack” [14] with about 1 in 20 suffering a stroke within the subsequent 48 h, presenting a golden opportunity for prevention of further brain ischemia [15]. The 2009 American Heart Association (AHA) scientific statement on the definition and evaluation of TIA adopted an imaging-based definition [16]; a change from the prior temporal-based definition. The statement formally defines TIA as “a transient episode of neurological dysfunction caused by focal brain, spinal cord, or retinal ischemia, without acute infarction.” The change in the definition was required because up to 50 % of TIAs diagnosed by the former definition have evidence of infarction on magnetic resonance imaging (MRI) (diffusion-weighted). TIA Class 1 recommendations of the AHA include imaging within 24 h, either MRI with diffusion weighting or computed tomography (CT)—though MRI is preferred. The proposed change in definition from time-based to imaging-based has implications for international clinical trials involving countries in which urgent MRI is not routinely available. Noninvasive imaging of the extracranial carotid arteries and intracranial cerebral vessels is also recommended.
Silent brain infarcts are a category of stroke in which stroke is evident on imaging studies in the absence of a history of TIA or stroke. The reported prevalence of silent brain infarcts is 8–28 % of patients in eight population-based studies [17]. Despite the lack of a history of TIA or stroke, these patients have been found to have neurological deficits including cognitive impairment, depression, visual field deficits, limb dysfunction, frailty, and physical decline. Thus, it has been suggested that they be referred to as “covert strokes.” Their prevalence is up to 5 times that of stroke and increases with age [17]. Covert brain infarcts detected by imaging are thus not necessarily asymptomatic. They are also not benign. Once detected, management implications are unclear and they are an important issue for ongoing stroke prevention research. Subclinical brain ischemia is potentially preventable, though there are no proven interventions to date. Definitive randomized controlled trials of current secondary preventive treatments are required to demonstrate the benefit of treatment in this patient population.
Clinically recognized strokes therefore represent the tip of the iceberg of vascular-mediated injury to the brain. A recently published prospective cohort study of the timing of onset of cognitive decline followed 10,308 British civil servants, performing tests of memory, reasoning, vocabulary, and semantic fluency; assessed 3 times over 10 years and found cognitive decline evident at ages 45–49 [18]. The Honolulu-Asia Aging study of 3,735 Japanese-American men showed that every 10 mmHg increase in midlife systolic pressure increased cognitive impairment by 7 % [19]. Whilst blood pressure lowering has not been shown to reduce vascular cognitive decline, there may be several reasons why this may be the case, including outcome measures that lack the sensitivity to detect appreciable differences when they are present.
Cardio-embolic stroke constitutes approximately 20 % of ischemic stroke and this proportion has remained relatively stable over the past few decades. This subtype of stroke is diagnosed when the probable cause of the stroke is presumed to be a consequence of embolism arising from the heart. The clinical diagnosis of cardio-embolic stroke can be made using the criteria outlined in Table 1.2.
Table 1.2
Criteria for the clinical diagnosis of cardio-embolic stroke
Identification of potential cardio-embolic source (stratified as major-risk vs. minor-risk) |
Absence of other likely causes of stroke |
Neurological features |
Demonstration of embolic occlusion (i.e., vanishing occlusions) |
Examples of major-risk sources include atrial fibrillation, intracardiac thrombus, atrial myxomas, and infective endocarditis. Minor-risk sources include calcific aortic stenosis, patent foramen ovale, mitral valve prolapse with myxomatous valves, and left ventricular systolic dysfunction with no associated thrombus (Figs. 1.3 and 1.4; Table 1.3).

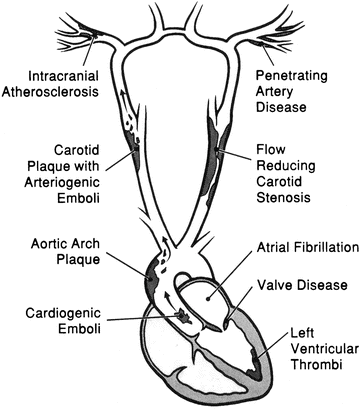
Fig. 1.3
Diagnosis of cardio-embolic stroke
Fig. 1.4
Embolic stroke mechanisms. Reprinted with permission from Robert G. Hart, M.D.
Table 1.3
Sources of cardiogenic brain embolism
Valvular diseases |
Rheumatic mitral stenosis |
Prosthetic valves |
Calcific aortic stenosis |
Mitral annulus calcification |
Marantic endocarditic and other prothrombotic states (e.g., antiphospholipid antibodies, diffuse intravascular coagulation, Wegener’s granulomatosis) causing valve thrombi |
Infective endocarditis |
Inflammatory valvulitis: Libman-Sacks endocarditis, Behcet’s disease, syphilitic |
Myxomatous mitral valvulopathy |
Left ventricular thrombi |
Due to ischemic heart disease: acute myocardial infarct, ventricular akinesis, or aneurysm |
Due to nonischemic, dilating cardiomyopathies: hypertrophic, amyloid, rheumatic myocarditis, neuromuscular disorders, catecholamine induced, doxorubicin, idiopathic, viral, peripartum, contusion, hypereosinophilic, sarcoid, alcoholic, Chaga’s disease, crack cocaine use, oxalosis, echinococcosis |
Left atrial thrombi without mitral valve disease |
Due to primary arrhythmias: atrial fibrillation, sick-sinus syndrome/atrial asystole, atrial flutter |
Due to other structural disease: atrial septal aneurysm, Chiari network |
Cardiac tumors |
Atrial myxoma |
Cardiac sarcoma |
Papillary fibroelastoma |
Metastatic |
Paradoxical emboli (trans-cardiac embolism) |
Atrial septal defects |
Patent foramen ovale |
Ventricular septal defects |
Pulmonary arteriovenous fistulas |
Miscellaneous |
Postcardiac catheterization
Related posts: Dysexecutive Syndrome After Stroke
Clinical Outcomes, Stroke Trials, and Cognitive Outcome Dysexecutive Syndrome After Stroke
Clinical Outcomes, Stroke Trials, and Cognitive Outcome
 Behavior After Aneurysmal Subarachnoid Hemorrhage: Cognition and Functional Outcome Behavior After Aneurysmal Subarachnoid Hemorrhage: Cognition and Functional Outcome
 Limb Apraxia: Types, Neural Correlates, and Implications for Clinical Assessment and Function in Daily Living Limb Apraxia: Types, Neural Correlates, and Implications for Clinical Assessment and Function in Daily Living
 Spatial Neglect: Not Simply Disordered Attention Spatial Neglect: Not Simply Disordered Attention
 Cognitive Dysfunction After Intracerebral Hemorrhage, Vasculitis, and Other Stroke Syndromes Cognitive Dysfunction After Intracerebral Hemorrhage, Vasculitis, and Other Stroke Syndromes
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|