Spreading Depression
Martin Lauritzen
Richard P. Kraig
The characteristic development of sensory disturbances during migraine auras suggests that the underlying mechanism is a disturbance of the neocortex, probably spreading depression of Leão (SD) (53). This viewpoint is supported by unique brain blood flow changes during migraine with aura, which are replicated in animal experiments during neocortical SD (47) (Fig. 28-1). Furthermore, SD in both the neocortex (55) and hippocampus (37) triggers trigeminal nucleus nociceptive activation, and so might also initiate migraine pain. The literature concerning SD is overwhelming, and a number of reviews cover most studies (4,9,47,57,72). The possible link between SD and migraine was reviewed recently (47) and is described here mainly in relation to newer studies.
DESCRIPTION OF THE PHENOMENON
Neurons and glia depolarize during SD, often associated with an initial transient of burst activity lasting seconds. This is followed by a complete loss of neuronal activity, which can last minutes before complete recovery occurs over several minutes (4). This sequence of brief excitation followed by a short-lasting depression is suggested to underlie the neurophysiologic basis of the sensory symptoms during migraine aura. SD is associated with a dramatic failure of brain ion homeostasis, efflux of excitatory amino acids from nerve cells, and enhanced energy metabolism (Fig. 28-2). SD has been induced in most gray matter regions studied, including neocortex, hippocampus, and cerebellum of a variety of species (4). Furthermore, human neocortical (50) and hippocampal (73) tissue in vivo and their counterparts studied in vitro (1,2) support SD. However, an electrophysiologic demonstration of SD in either neocortex or hippocampus during a migraine attack in humans is still missing.
HOW DOES SPREADING DEPRESSION SPREAD?
Several explanations for the spreading mechanism of SD have been proposed and likely involving reaction-diffusion processes (57). For example, elevated potassium and/or glutamate have long been suggested to be involved in SD initiation and propagation (57,60). Furthermore, SD in the neocortex of a variety of species, including humans, depends on activation of a single receptor, the NMDA receptor (1), one of the three subtypes of glutamate receptors (48). Calcium influx triggered by NMDA-receptor activation is the proper stimulus for nitric oxide synthase (NOS) activity, and part of the cerebrovascular response to SD depends on nitric oxide (NO). Several studies have shown that induction of SD by microdialysis of potassium or agonists for the NMDA receptor is associated with efflux of glutamate at the site of elicitation (59,68). During propagation of a SD, the interstitial concentrations of a number of amino acids, including glutamate, glycine, and taurine, increase and extracellular arginine decreases (15). The latter change may be relevant to the increased NO synthesis and release during and following SD as demonstrated recently (75) by an NO-sensitive microelectrode (65). Finally, SD triggers a profound release of cytokines whose reactivity there may contribute to SD propagation (40). Use of microdialysis could not determine whether massive release of transmitters occurred before or after the depolarization wave of SD because of low temporal resolution. Voltametry is more sensitive and shows that massive transmitter release follows the onset of the DC shift techniques (54,62). This suggests that profound chemical neurotransmitter release plays only a secondary role in the spreading mechanism of SD, and thus potassium takes center stage with a primary role. However, highly sensitive optical current source density analyses of SD suggest that more physiologic, yet elevated, levels of potassium and transmitters
may yet prove sufficient to trigger the spread of SD (41). Furthermore, initially rising interstitial potassium, by decreasing the resting membrane potential, might remove the voltage-sensitive Mg2+ block of the NMDA receptor, and so sensitize the receptor to small increases of interstitial glutamate. Then, receptor activation would trigger further neuronal depolarization and other biochemical events known to be associated with NMDA receptor activation, including potassium release.
may yet prove sufficient to trigger the spread of SD (41). Furthermore, initially rising interstitial potassium, by decreasing the resting membrane potential, might remove the voltage-sensitive Mg2+ block of the NMDA receptor, and so sensitize the receptor to small increases of interstitial glutamate. Then, receptor activation would trigger further neuronal depolarization and other biochemical events known to be associated with NMDA receptor activation, including potassium release.
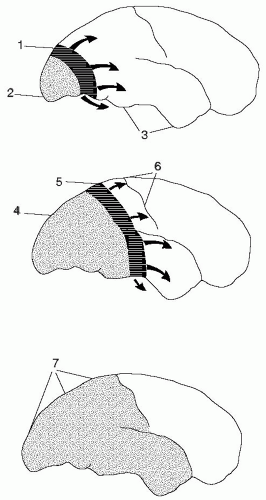 FIGURE 28-1. Hypothesis of development of attack of migraine with aura based on aspects of spreading depression (SD) and migraine. The figures represent lateral views of the human brain at different intervals after the beginning of the attack, spaced by approximately 30 minutes. Dotted area: Represents the region of reduced cerebral blood flow. Darkened area: The region of neuronal depolarization during the first minutes of SD. The direction of propagation of SD (arrows). (1) Initially during an attack of migraine with aura an SD is elicited at the occipital pole, spreading anteriorly at the lateral, mesial, and ventral sides of the brain. At the SD wave front, transient ionic and metabolic disequilibria trigger perturbed neuronal function, cerebral blood flow changes and neurological symptoms. (2) Following SD, cortical cerebral blood flow decreases by 20 to 30% for 2 to 6 hours. (3) Cerebral blood flow in regions not invaded by SD remains normal until encountered by SD. (4) The region of reduced cerebral blood flow expands as SD moves anteriorly. (5) Somatosensory symptoms from the extremities appear when the SD invades the primary sensory cortex at the postcentral gyrus. (6) SD usually stops on reaching the central sulcus, but in many patients it does not even propagate this far. The ventral spread of SD causes greatest activation of pain-sensitive fibers and headache. (7) Full-scale attack. The SD has stopped and is now detectable as a persistent reduction of cortical cerebral blood flow. At this time, the patient suffers from headache, but has no focal deficits. (From Lauritzen M. Cortical spreading depression as a putative migraine mechanism. Trends Neurosci. 1987;10:8-13, with permission.) |
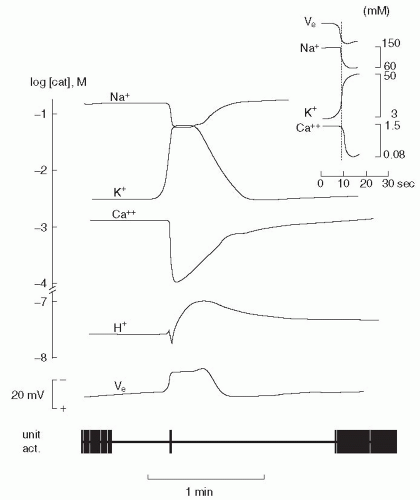 FIGURE 28-2. Electrophysiologic changes accompanying cortical spreading depression (SD) in the rat brain. Interstitial ion concentrations of sodium, potassium, calcium, and hydrogen were measured by ion-selective electrodes. Note that the scale is logarithmic. The extracellular potential (VE) and the single unit of activity were measured by single-barreled potential electrodes. SD was elicited in frontal cortex, and the electrophysiologic changes were recorded in the parietal cortex. The figure illustrates the dramatic failure of brain ion homeostasis during SD as sodium and calcium enter the cells while potassium and hydrogen ions enter the extracellular space. The disturbance is self-limiting, and after a few minutes, normal brain ion homeostasis is reestab-lished. (From Lauritzen M. Cortical spreading depression as a putative migraine mechanism. Trends Neurosci. 1987;10:8-13, with permission.) |
The importance of potassium fits well with recent data showing that the susceptibility of the tissue to produce SD is enhanced when the function of glial cells is hampered (44). Glial cells are major scavengers of interstitial potassium, and deterioration of glial cell function resulting from metabolic toxins leads to neuronal cell death in response to SD (44). Normal function of glial cells is important in
preventing SD initiation and neuronal damage during repeated episodes of SD, but normal glial cell function is not required for the SD to spread, because full-blown SDs can be produced while the glial cells are heavily poisoned, synaptic function being entirely normal (43). The glial depolarization during SD is related to passive Donnan-like KCl and water fluxes into astrocytes (76), which may have only a passive role in SD. However, neurons are likely to not only be responsible for the current signals initiating SD but also for those involved with it propagation and termination (41).
preventing SD initiation and neuronal damage during repeated episodes of SD, but normal glial cell function is not required for the SD to spread, because full-blown SDs can be produced while the glial cells are heavily poisoned, synaptic function being entirely normal (43). The glial depolarization during SD is related to passive Donnan-like KCl and water fluxes into astrocytes (76), which may have only a passive role in SD. However, neurons are likely to not only be responsible for the current signals initiating SD but also for those involved with it propagation and termination (41).
SD bears a strong resemblance to gap-junction-mediated calcium waves among cultured astrocytes (56). However, SD can propagate in nominally calcium-free media without evidence of these slower, glial-based waves (3). Nonetheless, more rapidly propagating neuronal calcium waves, presumably via gap junctions, are important for initiating SD (36). Furthermore, P/Q calcium-channel blockade can completely stop SD in hippocampus (38). SD in several in vitro preparations is sensitive to gap junctional blockade (36,49,56). Perhaps SD requires neuronal gap junctions and astrocytic gap junctions only facilitate the phenomenon. In addition, the sensitivity of hippocampal SD to P/Q calcium-channel blockade is not evident in neocortex (38). Taken together, these results suggest that new therapies for migraine based on gap-junctional or calcium-channel blockade need to be region and cell-type specific.
IS SPREADING DEPRESSION ONE OR SEVERAL PHENOMENA?
SD may develop differently, depending on the procedures used to elicit it. SD usually is recognized by its large interstitial DC potential change or by its corresponding interstitial potassium concentration change. Microinjection of potassium into the hippocampus can elicit a SD propagating in the pyramidal neuron dendritic layer with a faster onset, a lower threshold, a larger amplitude, and a faster propagation velocity than SD triggered in the pyramidal neuron cell-body layer. In some preparations, the direction of spread occurs in one direction in the dendritic layer and in the opposite direction in the cell-body layer (26). In rat cerebral cortex, the profiles of DC potential and potassium shifts change as a function of the depth and mode of elicitation (needle stab versus KCl application). Recurrent SDs propagating from the site of KCl application have longer duration and lower frequencies in superficial (dendritic) layers than in deeper (cell-body) layers. Potassium-induced SDs sometimes propagate independently in the superficial and deep layers. In contrast, SD elicited by needle stab usually invades the entire cortex with DC potentials that differ in shape from recurrent KCl-elicited SDs (66). Furthermore, there appears to be a barrier for the vertical spread of SD between a depth of 800 and 1,200 mm in neocortex. Thus, the electrophysiologic correlates of SD differ depending on the site of recording and the mode of elicitation in susceptible brain regions. Consequently, caution should be exercised and what is meant by SD always should be clearly defined when comparing results between groups.
Related posts:
 Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
Principles of Clinical Pharmacology, Randomized Controlled Clinical Trials, and Evidence-Based Medicine in Headache
 Nitric Oxide
Nitric Oxide
 Human Models of the Headaches
Human Models of the Headaches
 Channelopathies and Their Possible Relation to Migraines
Channelopathies and Their Possible Relation to Migraines
 Autonomic Dysfunction in Migraines
Autonomic Dysfunction in Migraines
 Psychological and Behavioral Treatments of Migraines
Psychological and Behavioral Treatments of Migraines
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


