Spinal Route of Analgesia: Opioids and Future Options for Spinal Analgesic Chemotherapy
Daniel B. Carr
Michael J. Cousins
Ignored until the late 19th century as a target for anesthesia, and for nearly another hundred years as a substrate for analgesia, the spinal cord has now emerged as a—if not the—key target for pain control in clinical anesthesiology. Indeed, the 1984 review of this topic by the senior author (1) has become the mostly frequently cited paper in the 20th century anesthesiology literature (2,3). In successive editions of this text, the topic of spinal analgesia has diffused outward from a small number of chapters, through many others that describe its application in acute pain, cancer pain, obstetrics, orthopedics, and pediatrics, to name but a few. More and more anesthesiologists now give drugs spinally to provide intraoperative (4,5) anesthesia and persistent postoperative analgesia after procedures as diverse as knee arthroscopy (Chapter 25), inguinal hernia repair (Chapter 23), or even (with a “light” general anesthetic) laparoscopic colectomy, thoracoscopy, or coronary artery bypass grafting (Chapter 22) (6). Such operations used to require extensive incisions under general anesthesia, with prolonged and painful hospital stays (7) and the risk of persistent postsurgical pain (8). Today, the combination of minimally invasive surgical techniques and spinal analgesia allows these procedures to be performed on a “fast-track,” often outpatient basis (9,10,11) with demonstrable benefit upon clinical outcome (12,13,14). Outpatient control of previously refractory cancer pain is now routinely accomplished by implanting pumps for spinal drug delivery (15,16,17,18). Intriguing evidence suggests that this approach to pain management may not only improve quality of life but also extend survival (19). These exciting advances harness sophisticated technologies to enhance patient outcomes and quality of life while lowering the cost (20,21,22) and burden of care. Such progress reflects the continuing translation of new knowledge about spinal analgesia discovered during the decade since the prior edition of this volume. In addition to the preclinical targets described in this chapter in the prior edition (23), newly elucidated mechanisms described by Yaksh in Chapter 32, and Dickenson and Seagrove in Chapter 33, offer promising new targets for spinal analgesia such as ion channels; receptors for a range of ligands, such as cannabinoids and vanilloids; trophic factors; and inflammatory mediators of systemic and local (e.g., glial) origin.
Balanced against these ongoing advances and prospects for spinal analgesia in acute and cancer pain are an emerging series of concerns that have tempered initial enthusiasm for intrathecal (IT) analgesia for chronic noncancer pain. As increasing numbers of patients have been treated for longer durations using implanted IT pumps, long-term risks and adverse effects, including loss of effectiveness, have become clearer. These negatives include drug-related effects (such as pedal edema or hypogonadism from opioids), possibly catastrophic pump- and catheter-related problems, and other issues such as infection or spinal headache (24). Particular concern has arisen about the inflammatory masses that may develop at the tip of catheters used to infuse opioids (25) and occasionally other drugs such as baclofen (26,27). First described in 1991 (28), such cases now approach 500. As they become more commonplace, new instances are less likely to be reported in the literature and more likely (albeit imperfectly) to be captured in registries. In light of a growing appreciation for the limits of chronic IT infusion, appropriate patient selection is increasingly important (see Appendix). However, the continuing dearth of randomized trials and clinical outcomes studies to inform such selection and subsequent management—a gap well noted in the prior edition published a decade ago—remains disappointingly large (29,30).
The present chapter begins with a brief summary of the history of spinal analgesia, supplementing earlier general historical chapters by Brown, and Gallagher and Fishman. We next survey relevant basic science, although not as comprehensively as Yaksh (Chapter 32), and Dickenson and Seagrove (Chapter 33), and others such as Walker (Chapter 30). The historical development of the modern approach to spinal analgesia has been described elsewhere by the senior author (31). We will then present highlights of the translation of preclinical advances into clinical practice, again in less detail than in other specific clinical chapters. Corresponding to the diffusion of spinal analgesia into many areas of regional anesthesia and pain medicine, it is no longer necessary for this chapter to cover each application of spinal analgesia in depth. Information relevant to pediatric spinal analgesia, for example, is provided in chapters on developmental aspects (Chapter 30), regional anesthesia (Chapter 27), and acute, chronic, and cancer pain (Chapters 43, 45,46,47). Cancer pain in adults (Chapter 45) is another valuable use of spinal analgesia, as may be complex regional pain syndromes (CRPS) and other forms of neuropathic pain. We will next survey ongoing efforts to translate preclinical insights into improved therapies, with emphasis upon acute pain and cancer pain. It is noteworthy that, in the decade since the prior edition, only one new medication specifically intended for IT use—ziconotide—has cleared all regulatory hurdles to enter the marketplace. Among trends in clinical practice, the use of “combination spinal analgesic chemotherapy,” a term introduced in the prior chapter and explored in detail in a subsequent systematic review (32), has burgeoned in the past
decade. Combinations of two or more agents coadministered intrathecally are now common, and in some current surveys account for the majority of chronic spinal infusion therapy. We will next discuss current and future prospects for combination therapy, mindful that (as Basbaum has pointed out in Chapter 51) clinical science now has in sight a day when the fundamental neurobiologic disturbances associated with persistent pain may be addressed in a mechanistic fashion, just as in other areas of biomedical science such as oncology or infectious disease. We will conclude with an illustrated approach to the implantation and management of spinal infusion therapy, including adverse events such as device failure or catheter-tip fibromas, whose prevalence has only begun to be fully appreciated. Although in the preparation of this chapter our broad literature search identified hundreds of thousands of publications related in some way to spinal analgesic therapy, the clinical trial literature on chronic spinal drug delivery has not matured sufficiently to support systematic reviews or meta-analyses. The persistent deficiency of high-quality evidence—clearly identified in the prior chapter as well as in the landmark 1984 paper by Cousins and Mather (1)—is a challenge that must urgently be faced, as regulators and third-party payers increasingly withhold support for therapies of unproven efficacy and cost effectiveness. Although lack of evidence does not prove the absence of benefit, particularly on an individual basis (33), failure to meet this challenge may well erode the clinical infrastructure necessary to sustain care and assure progress.
decade. Combinations of two or more agents coadministered intrathecally are now common, and in some current surveys account for the majority of chronic spinal infusion therapy. We will next discuss current and future prospects for combination therapy, mindful that (as Basbaum has pointed out in Chapter 51) clinical science now has in sight a day when the fundamental neurobiologic disturbances associated with persistent pain may be addressed in a mechanistic fashion, just as in other areas of biomedical science such as oncology or infectious disease. We will conclude with an illustrated approach to the implantation and management of spinal infusion therapy, including adverse events such as device failure or catheter-tip fibromas, whose prevalence has only begun to be fully appreciated. Although in the preparation of this chapter our broad literature search identified hundreds of thousands of publications related in some way to spinal analgesic therapy, the clinical trial literature on chronic spinal drug delivery has not matured sufficiently to support systematic reviews or meta-analyses. The persistent deficiency of high-quality evidence—clearly identified in the prior chapter as well as in the landmark 1984 paper by Cousins and Mather (1)—is a challenge that must urgently be faced, as regulators and third-party payers increasingly withhold support for therapies of unproven efficacy and cost effectiveness. Although lack of evidence does not prove the absence of benefit, particularly on an individual basis (33), failure to meet this challenge may well erode the clinical infrastructure necessary to sustain care and assure progress.
Overview
Only recently has the spinal cord taken “center stage” in analgesia practice and research. For thousands of years, pain relief could be secured only at the expense of central nervous system (CNS) depression, as with the use of mandragora, wine, and opium in ancient China; mandragora and “poppy” in ancient Egypt, Rome, and Greece; and atropine, opium, cocaine, and hallucinogens by the Incas and ancient Peruvians. Thus, until Koller’s daring introduction of local anesthetic blockade in 1884, the major site for pain control was thought to be the brain. In the early 20th century, the visionary surgeon Crile championed intraoperative neural blockade with the recently introduced local anesthetic cocaine used to avert “summation,” which we now recognized as sensitization, and to diminish the psychological and physiological sequelae of surgery, including persistent pain (34). The brain and axon continued to be the major options for analgesia (35) until the mid-1970s, when preclinical studies proved (36) that the spinal cord can be a target for selective opioid analgesia. By the early 1980s, with speed unprecedented since Koller’s time, these basic observations were harnessed in daily clinical practice worldwide (16,37,38,39,40). Insight into the chain of events precipitated in the spinal cord by painful peripheral stimuli has spurred novel drug discovery and rekindled interest in the spinal delivery of established drugs alone or in combination (32). This progress, milestones of which are summarized in Table 40-1, has been accelerated by the shared excitement of clinicians and investigators as they participated in a revolution of applied pharmacology. Patient-controlled analgesia (PCA) (41) first applied to the intravenous (IV) route in the 1960s, also was extended to spinal analgesia and, by the 1980s, was in wide use for epidural drug delivery.
Brain, spinal cord, axon, and periphery, once viewed as distinct sites for the actions of different analgesics, are now known to participate jointly as targets for pain control (42,43,44). Local anesthetics block axons of sensory (superficial) and motor tracts (deep) in the spinal cord (35). Systemically applied opioids and nonopioid compounds such as nonsteroidal anti-inflammatory drugs (NSAIDs) or clonidine have antinociceptive effects at the spinal cord level in animal studies and in humans (45,46). Opioids and NSAIDs also act to reduce pain and inflammation through direct effects upon peripheral tissue (47,48).
Epidural opioids can be absorbed rapidly into the circulation, producing early effects on the brain. Particularly hydrophilic (e.g., morphine) but also lipophilic opioids migrate in cerebrospinal fluid (CSF) to the brain, producing analgesia along with side effects such as sedation, dysphoria, nausea and vomiting, and respiratory depression. Very hydrophilic drugs, such as morphine and glucuronide metabolites, accumulate in the brain, to produce effects such as delayed respiratory depression. It is now clear that the spinal epidural administration of all opioids generates appreciable plasma concentrations that, for many drugs, approach the analgesic range (49,50,51). Only small amounts of opioid need reach supraspinal sites in order to augment spinal opioid analgesia (52,53,54). Hence, attempts to isolate the relative contributions of spinal, supraspinal (55,56), and systemic drug (and metabolite) actions during opioid therapy by any route may be irrelevant because all three sites of action are likely involved during treatment using all but the lowest doses and briefest courses.
Understanding of the spinal cord’s unique importance as a target for regional and systemic analgesics, reviewed in the prior editions of this text, continues to progress. Interim preclinical and clinical advances allow us to assess the effects of spinal analgesics across many scales of space and time, from interactions of ligand molecules and binding sites on specific receptors in dorsal horn neurons, to clinical outcomes that include patient satisfaction and cost of care. Without exaggeration, one may say that spinal cord sensitization has now replaced pituitary-adrenal activation (57) as the principal undesirable “stress response” whose suppression (or better, preemption) guides the clinical practice of perioperative regional anesthesia and analgesia. In contrast to classical stress responses quantitated as blood hormone levels (58), dorsal horn stress responses are intra- and interneuronal (e.g., remodeling) and manifest as long-term structural and functional changes after noxious input (59,60).
Opioids and Receptors: Terminology
The term narcotic (“narco” in Greek is to numb or deaden) is applicable to many drugs and is so vague that it is of little use except as a pejorative term in legal and regulatory contexts. The term opiate refers to morphine and related alkaloids derived from the poppy (some reports indicate that opiates are synthesized de novo in mammals, possibly by intestinal flora). Opioid, a broader term, includes exogenous substances with morphine-like properties as well as endogenous peptides (61). Endogenous opioid peptides or endorphins all have the aromatic amino acid tyrosine at the initial (i.e., amino or N-terminus) position; all opioids share this tyramine structural motif. Opioid peptides that lack this tyramine moiety do not exert morphine-like effects but still may possess other “non-opioid” biologic actions, such as enhancement of memory or immune modulation.
Full opioid agonists at the morphine or μ receptor include morphine, hydromorphone, meperidine, and fentanyl. These
agents bind to μ opioid receptors and activate them to produce dose-dependent analgesia and other effects (62) that are reversed by the opioid antagonist naloxone. As dose increases, there results a maximum or plateau of full efficacy. Some opioid agonists have less attraction to (affinity for) the binding site and require higher concentrations to achieve these effects. For other opioids, termed partial opioid agonists, this plateau is submaximal; buprenorphine is one example. Opioids that are agonists at one opioid receptor type and antagonists at a different opioid receptor type are termed, somewhat confusingly, mixed agonist-antagonists. Nalbuphine or butorphanol fall in this group; both are antagonists at the μ receptor and agonists at the κ receptor. Naloxone, a pure opioid antagonist, has high affinity and zero efficacy. The rate of dissociation of opioids from their receptors influences their duration of action. For example, buprenorphine has a slow rate of dissociation from the μ receptor and a long duration of action. For other drugs with a rapid rate of receptor dissociation, the concentration of opioid in the bloodstream or CSF, and drug redistribution and rate of clearance (e.g., by liver), govern duration of action (61).
agents bind to μ opioid receptors and activate them to produce dose-dependent analgesia and other effects (62) that are reversed by the opioid antagonist naloxone. As dose increases, there results a maximum or plateau of full efficacy. Some opioid agonists have less attraction to (affinity for) the binding site and require higher concentrations to achieve these effects. For other opioids, termed partial opioid agonists, this plateau is submaximal; buprenorphine is one example. Opioids that are agonists at one opioid receptor type and antagonists at a different opioid receptor type are termed, somewhat confusingly, mixed agonist-antagonists. Nalbuphine or butorphanol fall in this group; both are antagonists at the μ receptor and agonists at the κ receptor. Naloxone, a pure opioid antagonist, has high affinity and zero efficacy. The rate of dissociation of opioids from their receptors influences their duration of action. For example, buprenorphine has a slow rate of dissociation from the μ receptor and a long duration of action. For other drugs with a rapid rate of receptor dissociation, the concentration of opioid in the bloodstream or CSF, and drug redistribution and rate of clearance (e.g., by liver), govern duration of action (61).
Table 40-1 Milestones in spinal analgesic (particularly opioid) research | ||
|---|---|---|
|
The concept that cell membrane receptors mediate drug action arose in the 19th century. Descriptions of opioid structure–activity relationships that were presumed to reflect drug–receptor interactions appeared in the 1950s. Different types of opioid receptors were postulated in the 1960s by Martin, who, by the mid-1970s, had painstakingly catalogued heterogeneous profiles of the physiologic effects in dogs exposed to or withdrawn from a variety of opioids (63). Martin discerned three broad patterns of in vivo response to all opioids he evaluated and attributed these three profiles to selective activation of three distinct opioid receptor types. The first he termed “mu (μ),” for morphine; the second, “kappa (κ)”, for ketocyclazocine; and the third, “sigma (σ)”, for the proprietary drug SKF-10,047. The σ receptor is no longer considered as an opioid receptor; it is activated by phencyclidine. Soon after Martin published his results, Lord (64) and colleagues from the Kosterlitz group in Aberdeen postulated the existence of another opioid receptor type that they termed delta “(δ)” because of its identification in mouse vas deferens. An unusually long interval elapsed between pharmacologic identification of opioid receptor types in vivo and their cloning, reflecting technical obstacles such as the lack of a specific gene product to serve as a marker of opioid receptor activation and the typically inhibitory effect of such activation. All three types of opioid receptors—μ, κ, and δ—were cloned within a relatively short interval in the early 1990s (65).
Cloning of the opioid receptors confirmed and extended much information previously gleaned indirectly from pharmacologic studies (66). All three opioid receptor types are metabotropic; that is, coupled to guanyl nucleotide-binding regulatory proteins (G proteins) (67). They share structural
features with other G protein-coupled receptors including seven conserved hydrophobic domains that span the cell membrane, disulfide bonds between cysteine residues, glycosylation near the amino terminus, and phosphorylation by protein kinase A (PKA) in the first and third cytoplasmic loops as well as near the carboxy (C) terminus (68). Sequencing of DNA encoding these three receptors in rat and mouse reveals that each contains nearly 400 amino acids, and that all three types share substantial sequence homology not only with each other but also with receptors for somatostatin, angiotensin, and certain chemotactic factors. Messenger RNA for all three types of opioid receptors is present in dorsal root ganglia and superficial layers of the dorsal horn (69). Opioid binding to the μ receptor activates an intracellular G protein, Gi, that inhibits guanyl triphosphate formation (70). Pertussis toxin prevents Gi activation and is useful as a probe of Gi mediation of opioid effects in vivo and in vitro (71).
features with other G protein-coupled receptors including seven conserved hydrophobic domains that span the cell membrane, disulfide bonds between cysteine residues, glycosylation near the amino terminus, and phosphorylation by protein kinase A (PKA) in the first and third cytoplasmic loops as well as near the carboxy (C) terminus (68). Sequencing of DNA encoding these three receptors in rat and mouse reveals that each contains nearly 400 amino acids, and that all three types share substantial sequence homology not only with each other but also with receptors for somatostatin, angiotensin, and certain chemotactic factors. Messenger RNA for all three types of opioid receptors is present in dorsal root ganglia and superficial layers of the dorsal horn (69). Opioid binding to the μ receptor activates an intracellular G protein, Gi, that inhibits guanyl triphosphate formation (70). Pertussis toxin prevents Gi activation and is useful as a probe of Gi mediation of opioid effects in vivo and in vitro (71).
Perhaps the most exciting consequence of cloning the opioid receptors is the ongoing characterization of the genetic basis for variability of opioid responsiveness (72). Hundreds of reports within the past 5 years associate single nucleotide polymorphisms (SNPs) with individual differences in analgesia following systemic opioid administration. These SNPs affect not only the opioid receptor itself, but related processes including drug transport proteins (73) and enzyme systems such as catechol-O-methyl transferase (74). Gender, being chromosomally based, and race further influence analgesic responses to opioids (75). Principally, studies of the influence of SNPs upon opioid responsivity have involved systemic opioid administration, and the results are not uniform. Thus, much work remains to be done to elucidate the nature of genetic influences upon spinal mechanisms of opioid analgesia. Genetic influences upon nonopioid mechanisms of analgesia, such as sodium (Na) channels such as Na 1.7, are described by Seagrove and Dickenson (Chapter 33), as well as later in this chapter.
The straightforward mechanism of opioid receptor linkage to a G protein, and thence to intracellular effector mechanisms such as ion channels, does not do justice to the complexity of and ongoing controversy about the precise mechanisms by which opioids or other ligands interact with receptors in the cell membrane (76). Several proteins including adenylyl cyclases, G protein receptor kinases, β-arrestins, regulators of G protein signaling, and the scaffold protein spinophilin all play roles in opioid-related cellular signaling (77). These mediators are all involved in varied aspects of tolerance to opioids. Moreover, opioids and other peptides that bind to cell membranes contain helical regions that are amphipathic; that is, in which hydrophobic and hydrophilic amino acids are grouped on opposite faces of the helix. This structure positions the ligand, detergent-like, at the surface of the cell membrane (78). Partial immersion of the ligand’s amphipathic helical regions within the cell membrane can stabilize or destabilize the membrane independent of any specific ligand–receptor interaction (79). Further relevant to opioid pharmacology is the recognition that activation of G protein-coupled receptors results from a change in conformation from resting to active states, a “two-state” model first developed to describe the opening of transmitter-gated membrane ion channels (80). If a compound has a higher affinity for the conformation of the receptor in its active state, it is an agonist; if its affinity is equal for resting and active states, then it is an antagonist devoid of intrinsic activity; if its affinity is higher for the resting state, then it is an “inverse agonist,” capable of producing effects opposite from those of agonists. The relative affinity of a drug for active and resting states of the receptor also determines whether it will behave as a full or partial agonist.
Opioid Receptor Activation: Analgesic Effects in Vivo
Martin’s definition of opioid receptor types, which employed in vivo animal studies to extend and refine clinical observations, has remained the foundation for all subsequent preclinical and clinical studies in this area (81). During this time, efforts to characterize the receptor type selectivity of new opioids have shifted from whole-body pharmacologic studies toward in vitro methods that rely on mathematical analysis of drug–receptor affinity and drug displacement from receptor by highly selective reference opioids. At present, molecular methods to determine the primary sequence of opioid and other receptors, their chemical modification (glycosylation, phosphorylation, disulfide bridging) at specific sites, and the functions of particular subunits are well established. It is likely that the future literature on opioid receptors will continue this progressive shift in focus from bedside, to animal laboratory, to ligand–receptor binding, to molecular analysis of the receptor per se down to the level of single amino acid substitutions at ligand binding sites and other functional domains.
Unfortunately for clinicians practicing opioid analgesia, this paradigm shift has taken place unannounced (82). Therefore, the research literature is often confusing because different reports use distinct criteria to determine receptor selectivity of opioids (83,84). These diverse criteria include behavioral observation (e.g., animal performance in a Y-maze or lever-pressing as an index of subjective similarity between a test drug and a reference opioid); relative efficacy against experimental pain of different origins (e.g., heat versus pressure); potency compared with reference drugs in standardized in vitro bioassays (e.g., inhibition of electrical contraction of guinea pig ileum); comparison of dosage of nonselective (pA2) or selective antagonists to reverse a drug’s effects in vivo compared to effects of reference drugs; or quantitation of in vitro displacement of radiolabeled tracer amounts of test drug from receptor preparations or thin slices of tissue by increasing concentrations of unlabeled drug (Scatchard plot) or selective antagonists. These methods have made it clear that all three major types of opioid receptor—μ, κ, and δ— have subtypes, although information about subtype diversity has not yet had clinical application, and so will not be covered further in this chapter. Currently, preclinical determinations of novel opioids’ receptor selectivity rely upon in vitro measurements of their binding affinities for purified opioid receptors, concentrations of selective reference opioids necessary to displace them from defined receptors, and the effect of selective receptor alkylating agents in blocking their binding to receptors. On the other hand, the two-state and earlier models of ligand–receptor interaction dictate caution in extrapolating from binding data to inferences about receptor activation. For example, bremazocine binds equally to μ and κ receptors, but is selective in activating κ receptors. Therefore, at present, initial pharmacologic screening and characterization in vitro must be confirmed by testing in vivo of potency (e.g., ED50 for analgesia) and reversal of effect by selective opioid antagonists (85). This two-stage screening preserves the authenticity of in vivo observation yet spares much of its associated imprecision and minimizes animal use. Through this approach, considerable information has emerged on the opioid receptor modulation of analgesia and other clinically important processes such as cardiorespiratory, gastrointestinal, endocrine, and immune function.
Analgesia follows systemic or spinal administration of opioids that act upon μ, δ, or κ receptors (76,86). An important exception to this general rule is the phenomenon of
opioid-induced hyperalgesia (87) due to mobilization of excitatory neurotransmitter systems promptly upon opioid exposure (88,89). This phenomenon is described at greater length later. Other causes for hyperalgesia in animal models or case reports include accumulation of algesic metabolites such as morphine-3-glucuronide, opioid inhibition of spinal interneurons that are themselves inhibitory, or stimulation of the excitatory G protein (Gs) in preference to Gi at low opioid concentrations (90). The observation in knock-out mice lacking the μ receptor that high doses of morphine sufficient to activate δ receptors do not produce analgesia on tail flick testing indicates a unique role for the μ receptor in analgesia (91). Enhancement of analgesia (cooperativity), and in some reports synergy, occurs when more than one type of opioid receptor is activated simultaneously, such as μ plus δ, that may also form heterodimers. Analgesic enhancement may also be achieved by coadministration of an opioid with other analgesics that act at nonopioid receptors as agonists (adrenergic, cholinergic, etc.) or antagonists (to substance P, cholecystokinin, NMDA, cytokines, etc.) (92,93,94,95,96,97) that block ion channels (calcium, sodium, etc.), or that inhibit algesic enzyme systems (cyclooxygenase [COX], nitric oxide synthase [NOS], etc.) (98) (see Chapter 32). Cooperativity between opioids active upon distinct receptors, and opioids with nonopioid analgesic molecules, is evident in natural processes of endogenous analgesia. For example, a well-described “mismatch” occurs between receptor selectivity of the opioid molecules released within the spinal cord or into the peripheral circulation, and the opioid receptors that are occupied by these endorphins both adjacent (“paracrine” effect) and distant (“endocrine” effect) to their sites of release. Anatomic sites of opioid analgesic actions are likewise multiple and include supraspinal areas, the spinal cord, and injured tissue in the periphery. Therapeutic benefits therefore follow coadministration not only of different classes of analgesic to the same site (e.g., spinal cord) but also of analgesics to multiple sites at once.
opioid-induced hyperalgesia (87) due to mobilization of excitatory neurotransmitter systems promptly upon opioid exposure (88,89). This phenomenon is described at greater length later. Other causes for hyperalgesia in animal models or case reports include accumulation of algesic metabolites such as morphine-3-glucuronide, opioid inhibition of spinal interneurons that are themselves inhibitory, or stimulation of the excitatory G protein (Gs) in preference to Gi at low opioid concentrations (90). The observation in knock-out mice lacking the μ receptor that high doses of morphine sufficient to activate δ receptors do not produce analgesia on tail flick testing indicates a unique role for the μ receptor in analgesia (91). Enhancement of analgesia (cooperativity), and in some reports synergy, occurs when more than one type of opioid receptor is activated simultaneously, such as μ plus δ, that may also form heterodimers. Analgesic enhancement may also be achieved by coadministration of an opioid with other analgesics that act at nonopioid receptors as agonists (adrenergic, cholinergic, etc.) or antagonists (to substance P, cholecystokinin, NMDA, cytokines, etc.) (92,93,94,95,96,97) that block ion channels (calcium, sodium, etc.), or that inhibit algesic enzyme systems (cyclooxygenase [COX], nitric oxide synthase [NOS], etc.) (98) (see Chapter 32). Cooperativity between opioids active upon distinct receptors, and opioids with nonopioid analgesic molecules, is evident in natural processes of endogenous analgesia. For example, a well-described “mismatch” occurs between receptor selectivity of the opioid molecules released within the spinal cord or into the peripheral circulation, and the opioid receptors that are occupied by these endorphins both adjacent (“paracrine” effect) and distant (“endocrine” effect) to their sites of release. Anatomic sites of opioid analgesic actions are likewise multiple and include supraspinal areas, the spinal cord, and injured tissue in the periphery. Therapeutic benefits therefore follow coadministration not only of different classes of analgesic to the same site (e.g., spinal cord) but also of analgesics to multiple sites at once.
We are not accustomed to viewing the clinical practice of spinal opioid analgesia as involving coadministration of opioids to several sites simultaneously, but this undoubtedly occurs within hours of beginning such therapy. Systemically administered opioids rapidly reach spinal cord, brainstem, and brain. Epidural opioids are distributed into the bloodstream and reach periphery, brainstem, and brain in addition to their spinal target; IT opioids also are carried rostrally in CSF and, to a lesser degree, the peripheral circulation. Brief courses of clinical opioid therapy for routine postoperative pain control by any route last at least a few days, allowing ample time for equilibration and access to multiple anatomic sites rich in opioid receptors. Yet, in contrast to many preclinical reports describing in vivo responses to single doses of spinal or systemic opioids or nonopioid analgesics, relatively few studies detail the analgesic effects of repeated yet short-term opioid administration.
The physiology of nociception and analgesia resulting from actions of spinally (or systemically) administered opioids upon the dorsal horn has been presented in overview in the introductory section and in detail in Chapter 32 by Yaksh. Opioid suppression of excitatory neurotransmitter release (e.g., substance P, calcitonin gene-related peptide, neurokinin A) from C but not A fibers reflects μ, δ, or κ opioid inhibition of calcium channels on the former but not the latter neurons. In particular, opioids act selectively upon small C fibers (99). By contrast, analgesic concentrations of the analgesic peptide somatostatin inhibit calcium channels on large but not small nociceptive afferents. Postsynaptic effects of μ and δ opioids upon wide-dynamic-range (WDR) neurons are both direct, resulting from occupancy of postsynaptic opioid receptors coupled through Gi to potassium channels, and indirect, due to activation of inhibitory pathways that descend from the brainstem to the spinal level. Dual pre- and postsynaptic effects also result from activation of α2 and γ-aminobutyric acid (GABA) receptors, respectively, by selective agonists such as clonidine or baclofen. Additional opioid analgesic effects at the spinal level include stimulation of adenosine release and activation (i.e., disinhibition) of spinal interneurons, the latter contributing to opioid-induced pruritus. Despite similarities of receptor pharmacology in synapses within distinct nociceptive pathways of the dorsal horn (shown in Fig. 40-1), important preclinical and clinical differences exist between analgesic efficacy of specific drugs applied to treat pain of different origins. By the early 1980s it was clear that μ opioids are more potent than κ opioids against thermal pain, and that the reverse is true for pain of mechanical origin (e.g., pressure) (100). Since then, considerable evidence has accumulated that thermal hyperalgesia at the spinal level is mediated principally by activation of N-methyl-D-aspartate (NMDA) receptors, activation of protein kinase C (PKC), and generation of nitric oxide (NO) and cyclic guanosine monophosphate (cGMP) (101). Mechanical hyperalgesia relies principally on coactivation of spinal α-amino-3-hydroxy-5-methyl-4-isoxazopropionic acid (AMPA) and metabotropic glutamate receptors, activation of phospholipase A2, and the COX cascade (102). Other models of nociception such as peripheral nerve injury, inflammation from infectious or chemical agents, electrical shock, or application of toxins or irritants to the CNS each produce distinct pathophysiologies and display distinct dose–response profiles in potency testing of various opioid and nonopioid compounds. None of these models exactly duplicates common clinical pain problems such as acute postoperative pain, postcesarean pain, headache, low back pain or cancer pain—syndromes that each involve multiple known and unknown pain mechanisms acting in concert (103).
Tolerance to opioid analgesic effects refers to a decline in analgesic effect and/or the need to escalate opioid dosage during ongoing therapy (104). Opioid tolerance occurs in many preclinical pain models, most reliably in “normal” animals without pain that begin opioid treatment well before challenge with a nociceptive stimulus. This simple yet important phenomenon is complemented by an equally commonplace occurrence that animals or patients who have chronically received an opioid to control pain predictably experience “rebound” hyperalgesia when challenged with an opioid antagonist. Both observations imply that opioid tolerance is not—as believed for years and described in the 2nd edition of this volume—simply a passive consequence of loss of opioid receptors and/or decoupling of opioid–receptor binding from Gi activation (105). If this were all that happened, then challenge with an opioid antagonist might have little effect. Instead, the occurrence of these phenomena implies that opioid tolerance involves mobilization of active processes whose potential to produce latent hyperalgesia is masked so long as opioid dosing is maintained. The presence of these active processes does not mean that downregulation (decreased receptor number) and desensitization (decreased receptor–effector coupling) do not take place: they do (106). However, they cannot by themselves explain the everyday occurrences just mentioned. Thus our view of opioid tolerance has changed during the past 15 years from an analogy with passive muffling of an ongoing signal to one of active cancellation of the signal, followed by rebound of the now-unopposed countersignal when the original signal ceases (107).
Mechanisms of active cancellation of an ongoing opioid analgesic signal involve processes ranging from intracellular
and organ-level metabolic adaptations such as enhanced glucuronidation of morphine, to activation of anti-opioid, hyperalgesic peptide systems and glia. Activation of the μ opioid receptor results in translocation of intracellular PKC and phosphorylation of the calcium channel within the NMDA receptor complex (107). Phosphorylation of this channel removes its magnesium block and allows calcium entry just as after a nociceptive stimulus, even though nociception per se need not have occurred (108). Activation of NOS also mediates NMDA receptor effects, and co-administration of either a NOS inhibitor or an NMDA antagonist along with a μ opioid retards development of tolerance (60). Interestingly, tachyphylaxis to local anesthetic effects is also inhibited in a dose-dependent fashion by a NOS inhibitor or an NMDA antagonist (109). Animal studies have further demonstrated that tolerance develops more slowly when opioids with high intrinsic activity are administered, compared with equianalgesic doses of opioids with low intrinsic activity. Opioids of higher intrinsic activity require fewer receptors to be occupied to produce a response such as analgesia, thereby leaving their target cells with relatively more unoccupied “reserve” or “spare” receptors. Previously, the slower rate of tolerance developed during exposure to opioids of high intrinsic activity was identified with a higher number of remaining spare receptors. Given the connection (104,107) between μ receptor activation and intracellular NO and NMDA effects, a more plausible explanation of the slower development of tolerance during treatment with opioids of high intrinsic activity is that binding and activation of fewer μ receptors yields relatively less stimulation of intracellular NOS and PKC. Phosphorylation of the μ opioid receptor itself during prolonged activation has been proposed as an additional mechanism of opioid tolerance (67,110). In addition to intracellular mechanisms, anti-opioid peptides participate in opioid tolerance. In particular, activation of cholecystokinin and substance P synthesis and release oppose the effect of opioid analgesics, as inferred from augmentation of opioid analgesia after administration of antagonists to either hyperalgesic peptide (111,112).
and organ-level metabolic adaptations such as enhanced glucuronidation of morphine, to activation of anti-opioid, hyperalgesic peptide systems and glia. Activation of the μ opioid receptor results in translocation of intracellular PKC and phosphorylation of the calcium channel within the NMDA receptor complex (107). Phosphorylation of this channel removes its magnesium block and allows calcium entry just as after a nociceptive stimulus, even though nociception per se need not have occurred (108). Activation of NOS also mediates NMDA receptor effects, and co-administration of either a NOS inhibitor or an NMDA antagonist along with a μ opioid retards development of tolerance (60). Interestingly, tachyphylaxis to local anesthetic effects is also inhibited in a dose-dependent fashion by a NOS inhibitor or an NMDA antagonist (109). Animal studies have further demonstrated that tolerance develops more slowly when opioids with high intrinsic activity are administered, compared with equianalgesic doses of opioids with low intrinsic activity. Opioids of higher intrinsic activity require fewer receptors to be occupied to produce a response such as analgesia, thereby leaving their target cells with relatively more unoccupied “reserve” or “spare” receptors. Previously, the slower rate of tolerance developed during exposure to opioids of high intrinsic activity was identified with a higher number of remaining spare receptors. Given the connection (104,107) between μ receptor activation and intracellular NO and NMDA effects, a more plausible explanation of the slower development of tolerance during treatment with opioids of high intrinsic activity is that binding and activation of fewer μ receptors yields relatively less stimulation of intracellular NOS and PKC. Phosphorylation of the μ opioid receptor itself during prolonged activation has been proposed as an additional mechanism of opioid tolerance (67,110). In addition to intracellular mechanisms, anti-opioid peptides participate in opioid tolerance. In particular, activation of cholecystokinin and substance P synthesis and release oppose the effect of opioid analgesics, as inferred from augmentation of opioid analgesia after administration of antagonists to either hyperalgesic peptide (111,112).
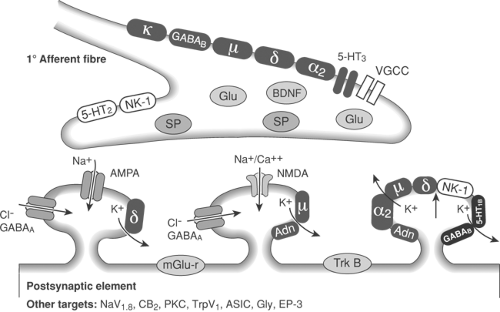 Figure 40-1. Schematic of pre and post–synaptic dorsal horn receptors and transmitters (see also Chapter 31, Fig. 31-11 and Chapter 32, Fig. 32-34). |
The contrast between the ease with which profound opioid tolerance can be induced during spinal or systemic infusions in many species of intact animals, and opioids’ sustained therapeutic value during chronic use in most clinical settings (113), is still not well understood. In the cat, tolerance to systemic administration of morphine can be demonstrated in dorsal horn neurons after 3 days (114). In the primate, daily IT administration of morphine, β-endorphin, or metkephamid, at doses producing a “just maximum effect” causes a daily reduction in analgesic efficacy. Responsiveness in the primate declines most rapidly for agents having the longest duration of action after each fixed dose. In mice, once-daily doses of opioids of differing intrinsic activity produce tolerance at the same rate, although continuous infusions elicit tolerance more quickly with opioids of low intrinsic activity (115). The latter result is also found during long-term spinal delivery of morphine, sufentanil, and alfentanil in dogs (51). Results from several species indicate that tolerance develops more quickly when a greater proportion of μ receptors is activated by ligand during a greater proportion of each day. Primates made tolerant to IT opioids began to recover opioid responsivity 7 days after the last intrathecal administration, and had near-complete recovery by 2 weeks, indicating that tolerance is reversible (116) and supporting clinicians’ common practice of managing tolerance to spinal opioid administration by switching temporarily to an alternative agent, such as spinal local anesthetic. It should be
emphasized that tolerance is a greater issue in intact animals or normal volunteers, than during opioid therapy of chronic experimental or clinical pain. Colpaert found that animals exposed to acute pain (mechanical pinch) or chronic pain (intraarticular Mycobacterium butyricum) failed to show tolerance to systemically administered opioids when compared with normal controls (117). Similarly, Glynn and Mather (118) reported that tolerance was not an inevitable consequence of prolonged (1 year) treatment of chronic pain in patients receiving systemically administered meperidine. Parallel observations emerged from surveys of IT opioid dose escalation in patients with pain from stable neoplasms by Foley (119), and patients with noncancer pain by Onofrio and Yaksh (120). In aggregate, observations by practitioners caring for patients with chronic pain from cancer or nonmalignant conditions confirm that systemic or spinal opioid dosage escalation is generally modest and therapeutic effectiveness is sustained, unless the underlying medical condition progresses (120,121).
emphasized that tolerance is a greater issue in intact animals or normal volunteers, than during opioid therapy of chronic experimental or clinical pain. Colpaert found that animals exposed to acute pain (mechanical pinch) or chronic pain (intraarticular Mycobacterium butyricum) failed to show tolerance to systemically administered opioids when compared with normal controls (117). Similarly, Glynn and Mather (118) reported that tolerance was not an inevitable consequence of prolonged (1 year) treatment of chronic pain in patients receiving systemically administered meperidine. Parallel observations emerged from surveys of IT opioid dose escalation in patients with pain from stable neoplasms by Foley (119), and patients with noncancer pain by Onofrio and Yaksh (120). In aggregate, observations by practitioners caring for patients with chronic pain from cancer or nonmalignant conditions confirm that systemic or spinal opioid dosage escalation is generally modest and therapeutic effectiveness is sustained, unless the underlying medical condition progresses (120,121).
In the authors’ view, rapid dosage escalation early during opioid therapy of chronic pain condition is evidence less of tolerance than of either a behavioral (psychological) issue, or of pain mechanism(s) that are intrinsically insensitive to opioid analgesia. In such cases, switching to a different opioid or nonopioid, or coadministering opioid plus nonopioid, are supported both by preclinical and clinical observations. Animals rendered tolerant to morphine by daily IT injections showed no loss of sensitivity to the δ opioid D-Ala(2), D-Leu(5) enkephalin (DADLE), and only partial loss of effect of the κ opioid ethylketocyclazocine (EKC) (122). Other studies have reported spinal analgesic effects with the κ agonist U-50-488H (123) and a loss of effect of the endogenous κ opioid dynorphin (a κ opioid) in animals made tolerant to EKC. The prospect of maintaining spinal opioid analgesia for prolonged periods by rotating opioid agonists of different receptor selectivity as tolerance develops has clinical potential, and is supported by preclinical and limited clinical studies (124). Some agents that act both at μ and δ receptors, such as metkephamid (125) or biphalin, are effective as analgesics in morphine-tolerant animals, as, too, are hybrid peptides whose structure has one part that functions as an opioid agonist and another that functions (counterintuitively) as a substance P–like agonist (126).
Nonopiate spinal analgesia has been extensively investigated in both animal and human studies and is becoming an increasingly prevalent component of clinical practice. In particular, the α-agonists (e.g., clonidine) have been shown to have powerful antinociceptive effects, which show no cross-tolerance with opioid agonists (116,127,128). Intrathecal and epidural clonidine have been successfully used in humans tolerant to spinal morphine (129), and the combination of clonidine and morphine is associated with sustained analgesic efficacy. In patients with “below level” neuropathic pain after spinal injury, spinal morphine is ineffective (130). However, in a rat model of spinal injury pain coadministration of IT morphine and clonidine does provide analgesia (131), and an initial report supports the clinical value of this drug combination for this purpose (132). Coadministration of catecholamines and enkephalin pentapeptides, secreted together from adrenal chromaffin cells implanted intraspinally, may be a future approach for prolonged analgesia (i.e., to avert tolerance), but early enthusiasm has waned owing to inconsistent results, the possibility of undetected infectious agents in xenotransplanted cells, the expense of this approach, and the potential for this experimental treatment to disqualify spinally injured participants from subsequent trials aimed at restoring spinal cord function per se. Concurrent infusion of spinal local anesthetic plus opioid also retains analgesic effectiveness for prolonged periods of time. Although the precise explanations for retarding tolerance by pairing opioid and nonopioid analgesic agents may vary, two common factors probably underlie this therapeutic advantage. First, addition of a second class of analgesic to an opioid reduces dorsal horn neuronal responsivity to afferent nociceptive input and hence lessens calcium influx, NOS activation, and PKC translocation. Second, insofar as coadministration of a second agent reduces the fraction of opioid receptors necessary to be activated in order to achieve analgesia, one would likewise expect tolerance to be retarded for the reasons just outlined. Thus, coadministration of opioids and other drugs, or administration of single molecules engineered to act upon opioid plus other pathways (e.g., COX, ion channels, substance P, NMDA, or NOS) is a powerful therapeutic strategy for achieving prolonged analgesia. Spinal antinociceptive systems that are potential targets for nonopioid analgesic molecules are summarized in Chapters 31,32,33. Their clinical relevance is discussed here.
Nonanalgesic Effects of Spinal Opioid Administration
The following summary describes the physiologic effects other than analgesia that directly result from spinal application of opioids. Accordingly, relationships between spinal opioid administration and alterations of respiratory, gastrointestinal, endocrine, cardiovascular, bladder, and sensorimotor function (the latter including pruritus and potential neurotoxicity and granuloma formation) are surveyed herein. Potential side effects of IT and epidural opioid treatment are summarized in Table 40-2 and contrasted with those of local anesthetics.
The present section is intended to complement the earlier discussion of the pharmacology of spinally administered opioid analgesics in preclinical and clinical settings, rather than to provide a comprehensive review of clinical effects, side effects, and outcomes other than analgesia during spinal opioid use. Such outcomes, which involve pulmonary or immune status, morbidity, or length of hospital stay, depend upon multiple processes including many not directly related to spinal opioid actions per se, such as are discussed later in this chapter and also by Liu and Wu in Chapter 7.
Respiratory Depression
Respiratory depression, although infrequent, is always a prime concern after spinal opioid administration, particularly in opioid-naive subjects treated for acute pain. Prompt (less than 2 hours) versus delayed onsets of this side effect have been attributed, respectively, to blood-borne drug quickly reaching the brain (such as after rapid absorption into the circulation of lipophilic opioid given epidurally) versus slow rostral migration of hydrophilic drug (such as morphine deposited within the CSF) (Fig. 40-2).
These two mechanisms are not mutually exclusive, however: sufentanil and meperidine have been detected in cisternal CSF within minutes after epidural or IT administration, respectively. Lipophilic opioids may rapidly cross the dura (Fig. 40-3) and then migrate in CSF to the brainstem (Fig. 40-2). Subacute respiratory depression during chronic use is also a concern. Respiratory depression was not a strong focus of initial animal experiments on opioid receptors or their endogenous ligands that followed the birth of this field in the early to mid 1970s. In large part, this lack of concern reflected the substantially higher
doses (on a mg/kg basis) for opioids to produce respiratory depression in laboratory rats, dogs, and other nonhuman species. The great importance of this and other side effects was soon appreciated once spinal opioid analgesia came into common clinical use in the late 1970s and stimulated detailed animal studies. This concern has, if anything, been strengthened by the increased number of respiratory “near-misses” and catastrophes associated with the concurrent growth in popularity of IV PCA (41,133).
doses (on a mg/kg basis) for opioids to produce respiratory depression in laboratory rats, dogs, and other nonhuman species. The great importance of this and other side effects was soon appreciated once spinal opioid analgesia came into common clinical use in the late 1970s and stimulated detailed animal studies. This concern has, if anything, been strengthened by the increased number of respiratory “near-misses” and catastrophes associated with the concurrent growth in popularity of IV PCA (41,133).
Table 40-2 Effects and side effects | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Animal studies provide strong evidence to connect respiratory depression to CSF concentrations of opioids. Data in baboons and in humans after epidural morphine administration indicate peak levels in CSF near the brain at about 3 hours (134,135,136). Studies in other species of cephalad migration of morphine after IT administration indicate a more rapid time course, with drug reaching the ventral brain after only 15 to 30 minutes and the respiratory center regions by 60 minutes (137). Opioid traveling cephalad in CSF also may stream against the intracranial CSF circulation to gain retrograde access to the fourth ventricle, with subsequent rapid access to respiratory centers. An alternative site of respiratory depression is a group of cells in the ventrolateral medulla, the nucleus ambiguus, and retroambigualis. These loci are involved in control of both inspiratory and expiratory motor output, but are less dense in opioid receptors than the subependymal nuclei in the floor of the fourth ventricle. In animal studies, direct injection of opioids into the fourth ventricle or into CSF of the ventral brainstem region results in a rapid (3–5 minutes) onset of respiratory depression, which is similar for both sites of injection (138). Thus, diffusion of the drug through the brain tissue to the respiratory centers in the brainstem appears to be rapid once the drug reaches this area in sufficient concentration. Once in the intracranial CSF, opioid removal may occur with great efficiency at the choroid plexus (139), which appears to act as a “cerebral kidney” for these substances (Fig. 40-2). On the other hand, evidence in animals and humans indicates that morphine may be biotransformed within the CNS to active metabolites such as morphine-6-glucuronide, which can have potent and prolonged respiratory depressant as well as analgesic effects (140). It is evident from animal and human studies that μ receptors have a predominant role in opioid-induced respiratory depression, although in some species such as dog, δ receptors are also important in this regard. Central injection of μ-selective opioids in animals reduces ventilation by slowing respiratory rate. Feuerstein and colleagues have attributed the paradoxical effects of a variety of μ-selective opioids, some of which stimulate and others of which depress ventilation, to opposite effects of activation of μ1 and μ2 receptor subtypes. Activation of high-affinity μ1 receptors either by low doses of opioids such as morphine, or by selective μ1 agonists, appears
to stimulate ventilation, whereas activation of low-affinity μ2 receptors by higher opioid doses depresses ventilation. Interestingly, in animal species such as rat, μ1 receptors develop later than μ2 receptors, raising the possibility that relatively depressed numbers of μ1 receptors may underlie neonatal sensitivity to opioid-induced respiratory depression.
to stimulate ventilation, whereas activation of low-affinity μ2 receptors by higher opioid doses depresses ventilation. Interestingly, in animal species such as rat, μ1 receptors develop later than μ2 receptors, raising the possibility that relatively depressed numbers of μ1 receptors may underlie neonatal sensitivity to opioid-induced respiratory depression.
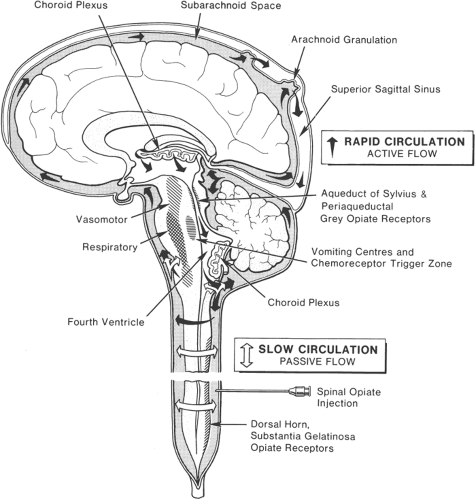 Figure 40-2. Model of CSF flow and spread of opioid in CSF. After lumbar intrathecal injection, opioid is carried in the passive flow of CSF to reach peak concentrations in the brain after about 3 hours for morphine and 0.5 to 1 hour for meperidine (Demerol). Rapid spread ensues when the opioid mixes with the active flow of the rapid circulation of intracranial CSF. Spinal and brain stem opioid receptors are shown. The latter are seen to be in proximity to cardiorespiratory and vomiting control centres. |
Intrathecal opioid, if given clinically as a single excessive dose or supplemented with IV opioid, may result in sudden apnea, necessitating rapid treatment. Onset of respiratory depression after IT morphine administration is variable (137,141,142,143,144,145,146,147,148) but usually is evident within 6 to 10 hours after the opioid injection. Return of normal respiration has required up to 23 hours afterwards. Case reports of respiratory depression when opioids were injected in usual doses intramuscularly within 24 hours of IT opioid (147) therefore, are not unexpected. Usually the progression of respiratory depression and hypercarbia after IT morphine is gradual, allowing time for diagnosis and treatment to avert respiratory arrest. This slow, insidious depression of ventilatory response to carbon dioxide (CO2) may be followed by sudden apnea, particularly when other risk factors are present, such as concomitant use of CNS depressants. In contrast, local anesthetic–induced convulsions or circulatory depression are usually rapid in onset and mandate urgent treatment.
Delayed respiratory depression after IT morphine for postoperative pain was first reported toward the end of 1979 independently by Glynn and associates (144) and by Liolios and Anderson (146). Glynn and colleagues (144) described two cases of respiratory depression persisting to 18 hours after a single dose of 3 mg and 5 mg of morphine, respectively. The patients were admitted to an intensive care unit and treated with repeated doses of naloxone. High doses of morphine (20 mg) were injected in a hyperbaric solution of dextrose by Samii and co-workers (149) with a similar duration of analgesia to that reported by Wang and others (150). Samii’s patients were nursed in a semi-sitting position, and side effects were not noted.
However, Liolios and Anderson (146), using a hyperbaric solution of 15 mg of morphine, did observe respiratory depression. Early in 1980, Davies and colleagues reported delayed and prolonged postoperative respiratory depression in three patients who received 1 mg of morphine in an isobaric solution (141,142). All three patients had been premedicated with a long half-life sedative, diazepam, and remained supine postoperatively. In retrospect, it seems likely that the lack of respiratory depression in initial clinical reports by Cousins and others (39,144), Wang and others (150), and Sami and others (149) reflected opioid tolerance, since their patients all had prolonged prior treatment with opioids for cancer pain. In contrast, the cases reported by Glynn and others (144) and Davies and others (141,142) were postoperative and opioid-naive. Lumbar IT injection of radionuclides (151,152) or water-soluble contrast media (153) (metrizamide) is followed by a gradual movement of these substances rostrally to reach the fourth and lateral ventricles after 3 to 6 hours. Contrast media can be demonstrated in the fourth and lateral ventricles 6 hours after lumbar injection, indicating major reflux into the ventricles by way of the foramina of Luschka. Gustafsson and associates found that [11C]-morphine appeared at a high cervical level after 60 to 170 minutes (137). Contrast also appears 6 hours after lumbar injection in the IT space over the entire surface of the brain and shows significant penetration of brain tissue (154,155). In summary, small volumes of opioid injected slowly into the IT space are likely to follow the passive circulation of CSF to reach the cisterns of the brain and then reach the respiratory center by way of the ventral pons (Fig. 40-2).
However, Liolios and Anderson (146), using a hyperbaric solution of 15 mg of morphine, did observe respiratory depression. Early in 1980, Davies and colleagues reported delayed and prolonged postoperative respiratory depression in three patients who received 1 mg of morphine in an isobaric solution (141,142). All three patients had been premedicated with a long half-life sedative, diazepam, and remained supine postoperatively. In retrospect, it seems likely that the lack of respiratory depression in initial clinical reports by Cousins and others (39,144), Wang and others (150), and Sami and others (149) reflected opioid tolerance, since their patients all had prolonged prior treatment with opioids for cancer pain. In contrast, the cases reported by Glynn and others (144) and Davies and others (141,142) were postoperative and opioid-naive. Lumbar IT injection of radionuclides (151,152) or water-soluble contrast media (153) (metrizamide) is followed by a gradual movement of these substances rostrally to reach the fourth and lateral ventricles after 3 to 6 hours. Contrast media can be demonstrated in the fourth and lateral ventricles 6 hours after lumbar injection, indicating major reflux into the ventricles by way of the foramina of Luschka. Gustafsson and associates found that [11C]-morphine appeared at a high cervical level after 60 to 170 minutes (137). Contrast also appears 6 hours after lumbar injection in the IT space over the entire surface of the brain and shows significant penetration of brain tissue (154,155). In summary, small volumes of opioid injected slowly into the IT space are likely to follow the passive circulation of CSF to reach the cisterns of the brain and then reach the respiratory center by way of the ventral pons (Fig. 40-2).
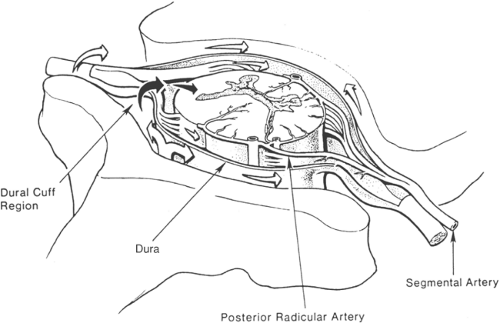 Figure 40-3. Cross section of spinal cord and epidural space. Opioid spread in epidural space is depicted by white arrows, and spread into CSF, and spinal cord, is depicted by black arrows. In dural cuff region, posterior radicular spinal artery is readily accessible to opioid, and this artery directly supplies the dorsal horn region of the spinal cord. |
Risk factors for respiratory depression after IT opioid administration include advanced age (143); poor general condition (156); use of water-soluble opioid (i.e., morphine) (137); high doses (144); marked changes in thoracoabdominal pressure, including artificial ventilation (143); lack of tolerance to opioids (144); and concomitant administration by other routes of opioids or other CNS depressant drugs (142,147). Patients with respiratory disease would be expected to be at risk, as they are with the use of opioids by any route. Age may influence spinal fluid volume and pressure, and the brain in elderly patients may be more susceptible to respiratory depression by opioids (157). The onset and offset of respiratory depression are in agreement with the time courses of minute volume and carbon dioxide response after epidural morphine in volunteers (Fig. 40-4). In many case reports, antagonism of respiratory depression by naloxone has been confirmed, but often several doses or an infusion of naloxone have been required to sustain this antagonism (141,144). Analgesia can be preserved during reversal of spinal opioid-induced respiratory depression by carefully titrating the doses or infusion rate of naloxone (145,156). It has been claimed that the use of the sitting position and a hyperbaric solution of morphine protects against respiratory depression (148). However, respiratory depression has been reported with such maneuvers, and so it seems wiser to limit the dose of drug rather than to rely on the sitting position (156).
Epidural opioid administration may also produce early or delayed respiratory depression. Compared with the IT route, epidural administration is complicated by pharmacokinetic aspects related to dural penetration, fat deposition, and systemic absorption. Pharmacodynamic aspects become complicated, since the larger doses of opioid used result in blood concentrations that cannot be ignored. Simultaneous lumbar epidural injection of morphine and meperidine in patients results in detectable levels of both drugs in cervical CSF at about 30 minutes after injection. Thereafter, CSF meperidine concentrations in the cervical region declines rapidly. The commercial approval of an epidural depot preparation of morphine encapsulated within microspheres of plastic “foam” has introduced another risk factor for prolonged hypoventilation into clinical practice (158).
Most reports of early respiratory depression after epidural meperidine have been in postoperative patients within 1 hour of injection (159,160) and probably reflect vascular absorption by way of epidural veins or possibly rapid redirection to brain by way of the basivertebral venous system (see below and Fig. 40-5A).
Thus, it is plausible that lipid-soluble drugs may cause early respiratory depression at least partly as a result of rapid access to the brain, to achieve peak concentrations near the brainstem at about 30 minutes. This concept is supported by studies of plasma concentrations and respiratory effects of fentanyl: plasma fentanyl concentrations peak after 5 minutes and then
decline rapidly, whereas respiratory depression is seen between 15 and 60 minutes (161,162). These pharmacokinetic considerations are consistent with clinical observations that delayed respiratory depression is most common with epidural morphine (163,164,165,166,167) and absent with epidural fentanyl (161,168). As in the case with IT opioid administration, old age, poor general condition, and respiratory disease probably predispose to respiratory depression (156,163,168,169). Patients subject to Stokes-Adams attacks also require caution (171). Finally, postoperative patients have a number of potential factors that place them at risk for this complication (Table 40-3).
decline rapidly, whereas respiratory depression is seen between 15 and 60 minutes (161,162). These pharmacokinetic considerations are consistent with clinical observations that delayed respiratory depression is most common with epidural morphine (163,164,165,166,167) and absent with epidural fentanyl (161,168). As in the case with IT opioid administration, old age, poor general condition, and respiratory disease probably predispose to respiratory depression (156,163,168,169). Patients subject to Stokes-Adams attacks also require caution (171). Finally, postoperative patients have a number of potential factors that place them at risk for this complication (Table 40-3).
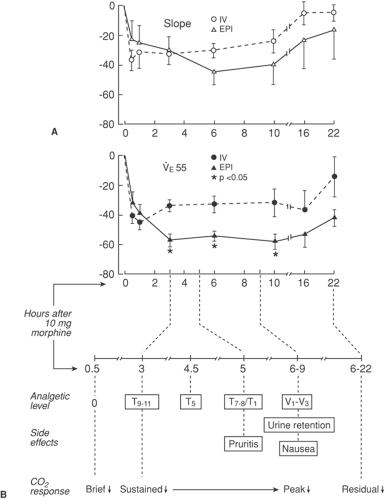 Figure 40-4. A: Ventilatory response to endogenous carbon dioxide CO2 after epidural (Epi) and IV morphine 10 mg by separate injection 2 to 4 weeks apart in 6 subjects. Mean ± SEM percentage change in slope and ventilation at end-tidal PCO2 of 55 mm Hg (VE55). (Reprinted from Neilsen CH, Camporesi EM, Bromage PR et al: CO2 sensitivity after epidural and IV morphine. Anesthesiology 1981;55:A372 with permission of the publisher). B: Respiratory and other side-effects following epidural morphine. |
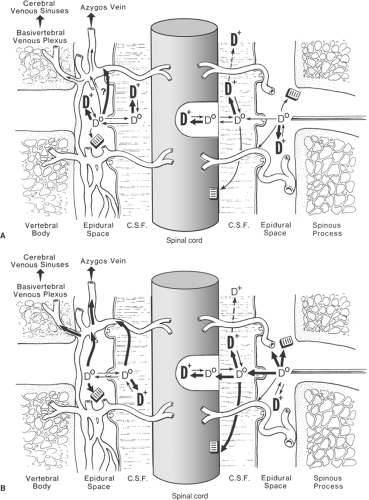 Figure 40-5. A: Pharmacokinetic model: Epidural injection of a hydrophilic opioid such as morphine. D0, Un-Ionized drug; D+, ionized hydrophilic drug. Major route of clearance is through CSF. Shaded squares are non-specific lipid binding sites. Role of radicular arteries in drug absorption remains unproven. B: Epidural injection of a lipophilic opioid such as meperidine or fentanyl. Note rapid passage of un-ionized species (D0) via CSF into cord and then clearance by epidural veins, leaving less ionized species (D+) remaining in CSF to migrate to brain. |
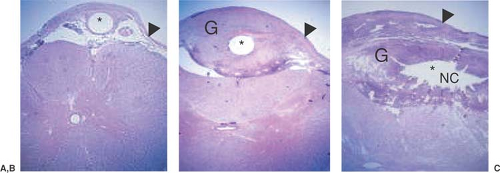 Figure 40-6. Spinal catheter-tip fibroma. Histopathology through upper lumbar (A) to caudal lumbar (B, C) spinal cord of a dog receiving intrathecal infusion of morphine (12.5 mg/mL at 40 μL/hr) for 28 days showing the rostrocaudal development of the granuloma from the meninges. Note the necrotic center in the largest area of the mass proximal to the catheter. This pathology is uniformly negative for infectious processes. Abbreviations: Arrowhead, Dura; G, granuloma; NC, necrotic center;* Catheter lumen. (This figure kindly provided by Prof. T Yaksh) [For radiological camparison see Chapter 50, Fig. 50-19]. |
Table 40-3 Factors potentially contributing to delayed respiratory depression during postoperative epidural opioid analgesia | |
|---|---|
|
Studies in volunteers by Bromage and others (172,173,174) suggested that changes in CO2 responsivity after epidural morphine paralleled its rostral spread, as judged by the cephalad progression of analgesia to ice and pin scratch (Fig. 40-4).
The concurrence of analgesia in the trigeminal distribution, nausea, and peak respiratory depression between 6 to 9 hours after epidural drug administration is strong evidence that significant brain opioid concentrations are reached during this interval. There appears to be a relatively slow progression of analgesia as morphine is carried in the passive flow of the slow circulation of spinal CSF and then merges with the active intracerebral CSF circulation (Figs. 40-2 and 40-3). This time course is in keeping with studies by Gourlay and associates (135), who reported peak morphine concentrations in cervical CSF 3 hours after lumbar epidural administration, as well as animal pharmacokinetic data (137), as described earlier.
Studies by Bromage and others (172,173,174) and Gourlay and colleagues (135) help in understanding the time delay of approximately 3 to 12 hours in clinical case reports of delayed respiratory depression after epidural morphine (166,167,175). From this work, it may be surmised that further systemic injection of “usual” doses of opioid would be dangerous for up to a day after epidural morphine. Indeed, this risk can be gauged from the 40% reduction in slope and ventilation after 10 mg of IV morphine, CO2-response beyond the residual 20% reduction in slope and 40% reduction in ventilation remaining at 22 hours after epidural morphine (173). A similar delay in onset of respiratory depression is seen after IT morphine (141,144,146,156), supporting the importance of the rostral spread of morphine in CSF. In an effort to exploit the effect of posture to inhibit rostral morphine spread, a “between-patient” study found the sitting position to be associated with less respiratory depression than the supine position after epidural morphine (176). However, a controlled study showed no effect of 45-degree elevated posture in protecting against respiratory depression (177). Use of a minimal effective dose of morphine is a more practical approach to achieving safety, since respiratory depression is dose-related (161,178). Doses of 2 to 4 mg morphine result in much milder and briefer depression of respiration than a dose of 10 mg morphine (161,178), which continues to depress respiration for more than 17 hours (178). Depot preparations of morphine present at least as great a duration of ventilatory risk (158). Fortunately, 2 to 4 mg of epidural morphine is often adequate for peripheral surgery, and 4 to 6 mg is adequate for more extensive surgery, provided sufficient time is allowed for onset (161,179,180). A wider therapeutic window can be obtained between analgesia and respiratory depression by using low-dose epidural morphine infusion preceded by a bolus dose in a low volume (1 mL), so that peak concentrations of morphine in brainstem regions are decreased and the incidence of respiratory depression is low (181,182,183). Respiratory depression can be prevented, or antagonized when it occurs, by naloxone infusion at a dose of 5 μg/kg/hr, without antagonizing analgesia (184). Epidural application of the partial μ agonist buprenorphine, and the mixed κ agonist–μ antagonist butorphanol have both been reported to induce respiratory depression for 12 hours after administration (1,185). The moral is clear: respiration must be carefully assessed if parenteral opioid is to be given within 24 hours of any epidural opioid, and parenteral dosage must be kept as low as feasible, preferably through cautious IV titration while an opioid antagonist is nearby.
All of the lipophilic opioids are reported to induce a brief period of early respiratory depression after epidural administration (186,187,188), possibly from blood-borne access to the brain but more likely as a result of transient peak concentration of drug in cisternal CSF (137,159) (see Fig. 40-2). Because most lipophilic opioids move rapidly into and out of neural tissue, delayed respiratory depression is exceedingly rare (162,189). These theoretical considerations are borne out clinically, provided the dose of fentanyl analogue is kept within the therapeutic margin of about 2, between safe epidural dose and analgesic intramuscular dose. Ventilatory effects of analgesic epidural doses of fentanyl, alfentanil, and sufentanil given to normal volunteers in a dose-ranging study resolved approximately 3 hours after drug administration (50). Also in volunteers, epidural fentanyl did not alter CO2 response curves over a 24-hour period of study, apart from a brief early depression (167). Low-dose epidural fentanyl infusion (0.5 mcg/kg/h) after a bolus of 1.5 μg/kg was not associated with changes in continuously monitored end-tidal CO2 or respiratory rate over an 18-hour monitoring period in 21 postoperative patients (161). On the other hand, epidural infusion of 1 μg/kg/hr fentanyl after a bolus dose of 1 μg/kg did depress CO2 responsivity throughout an 18-hour study period in patients after orthopedic surgery.
Extensive preclinical and clinical attention to spinal opioid–induced respiratory depression is offset by voluminous surveillance data attesting to the low incidence of this complication
in tens of thousands of patients treated with IT and epidural opioids. Large-scale surveys in Sweden and elsewhere (57,156,190) have enrolled nearly 30,000 patients and yield incidence estimates of respiratory depression after epidural morphine that range from 0.09% to 0.25%. These estimates compare favorably with published incidence rates for respiratory depression after parenteral morphine, whether given by conventional injection or PCA (39,133). Such surveillance also identified the rarity of respiratory depression occurring more than 12 hours after single doses of epidural morphine, and led the Swedish Society of Anesthesiology and Intensive Care to endorse routine observation for only 12 hours. Although few would question multiple experimental demonstrations of respiratory effects persisting beyond 12 hours in volunteers given epidural morphine, the interpretation of such responses to exogenous CO2 and their generalizability to patient care is not straightforward. For example, a variety of studies have found benefits of epidural opioid analgesia upon postoperative pulmonary function and oxygenation, presumably derived from enhanced pain control during deep breathing or coughing. Moreover, respiratory depression is recognized to be uncommon in patients who already are opioid tolerant (18,120,191,192), and there have been few reports of such in patients with chronic cancer-related or nonmalignant pain treated with long-term spinal opioids (191,193,194,195,196). A heightened risk of epidural opioid use occurs in patients with known or undiagnosed obstructive sleep apnea, in which a small epidural dose of morphine (or even parenteral morphine) can result in severe sedation and respiratory depression (197).
in tens of thousands of patients treated with IT and epidural opioids. Large-scale surveys in Sweden and elsewhere (57,156,190) have enrolled nearly 30,000 patients and yield incidence estimates of respiratory depression after epidural morphine that range from 0.09% to 0.25%. These estimates compare favorably with published incidence rates for respiratory depression after parenteral morphine, whether given by conventional injection or PCA (39,133). Such surveillance also identified the rarity of respiratory depression occurring more than 12 hours after single doses of epidural morphine, and led the Swedish Society of Anesthesiology and Intensive Care to endorse routine observation for only 12 hours. Although few would question multiple experimental demonstrations of respiratory effects persisting beyond 12 hours in volunteers given epidural morphine, the interpretation of such responses to exogenous CO2 and their generalizability to patient care is not straightforward. For example, a variety of studies have found benefits of epidural opioid analgesia upon postoperative pulmonary function and oxygenation, presumably derived from enhanced pain control during deep breathing or coughing. Moreover, respiratory depression is recognized to be uncommon in patients who already are opioid tolerant (18,120,191,192), and there have been few reports of such in patients with chronic cancer-related or nonmalignant pain treated with long-term spinal opioids (191,193,194,195,196). A heightened risk of epidural opioid use occurs in patients with known or undiagnosed obstructive sleep apnea, in which a small epidural dose of morphine (or even parenteral morphine) can result in severe sedation and respiratory depression (197).
Gastrointestinal Function
Most preclinical studies of the gastrointestinal effects of opioids in vivo have employed transit (propulsion) as an end-point, although some have examined mucosal transport of fluid and ions. These two physiologic effects are clearly separable: in some models, the antidiarrheal effects of opioids result principally from antisecretory rather than antipropulsive effects. The relationship between opioid (or other drug) effects upon gastrointestinal contractions (i.e., motility) versus propulsion (i.e., transit) is not a simple one, nor is it the same at different sites along the gut. For example, opioids delay propulsion in species in which they inhibit contractions; in other species, opioids increase contractions but likewise delay propulsion. Opioids can affect intestinal function (including gastric acid secretion) by actions in the brain, spinal cord, and periphery. Opioid μ agonists given in analgesic doses at any of these three sites inhibit propulsion, gastric acid secretion, and diarrhea. Analgesic doses of κ opioid agonists administered at any of these three sites have little effect upon propulsion or gastric acid secretion, and may even stimulate the latter process when given intravenously. Analgesic doses of δ opioid agonists given intracerebroventricularly or systemically do not affect propulsion or gastric acid secretion, but do inhibit diarrhea. Spinally administered δ opioid agonists, however, inhibit propulsion in preclinical models.
In patients followed postoperatively, gastrointestinal mobility normally decreases, particularly after abdominal surgery. In normal volunteers, opioids given systemically or epidurally delay gastric emptying and decrease gastrointestinal motility. However, one controlled trial in obese patients found a decrease in times to pass flatus and feces postoperatively, and shortened hospital stay, in those given epidural morphine compared with those given systemic (intramuscular) morphine (185). Gastrointestinal effects of postoperative epidural local anesthetics alone are minimal; the benefits of their use postoperatively are described in Chapter 7. Thus, one may provisionally conclude that clinical findings during application of spinal μ opioid agonists are consistent with preclinical effects, in particular that epidural μ opioid administration results in a slowing of postoperative recovery of gastrointestinal function intermediate between systemic opioid administration and spinal analgesia with local anesthetics. A comprehensive picture of the gastrointestinal effects of acute and chronic spinal opioid use in humans, that encompasses non-μ opioids, and coadministration of opioid-sparing analgesics, such as local anesthetics or nonsteroidals (198), has not yet emerged. Recognition of the detrimental effects of opioids upon gut mobility during both acute and chronic administration has prompted the development of peripherally active opioid antagonists (199) such as methylnaltrexone (200,201) and alvimopan (202,203). Nearly all experience with these and other novel agents has been acquired during systemic opioid therapy. As noted earlier, however, there is a significant contribution of central mechanisms to gut hypomotility following spinal opioid administration. Therefore, these and other peripherally active opioid antagonists now under development merit evaluation during spinal opioid analgesia.
Nausea and vomiting occur in approximately one-quarter of postoperative patients treated with spinal opioids, although pain itself has been implicated as a cause of postoperative nausea (180). In a prospective study of 1,085 postoperative patients, Stenseth and associates (180) reported that epidural morphine (dose 4–6 mg) was associated with nausea or vomiting in 34% of patients. In a series of 1,200 postoperative patients, nausea and vomiting were present in 17% of the patients (190), whereas in another series, nausea was present alone in 12%, and with vomiting in 24% (179). In the latter series, the incidence of nausea and vomiting was similar whether morphine was used intramuscularly or epidurally, or saline was injected epidurally. Others, too, have reported a low incidence of nausea and vomiting after epidural morphine in the postoperative period (204). In labor, the incidence of nausea and vomiting has been reported to be low with epidural opioid (186,205) in contrast to a high incidence with IT opioid (206,207). Epidural use of lipid-soluble opioids such as meperidine, fentanyl, and sufentanil (189,208,209) may be associated with the lowest incidence of nausea and vomiting. In a cross-over study in volunteers, Bromage and colleagues (210) observed nausea and vomiting in 50% of subjects approximately 6 hours after epidural morphine, which coincided with other evidence of the rostral spread of morphine in CSF to intracerebral structures, including the vomiting center and the chemoreceptor trigger zone (Fig. 40-4). In contrast, after IV morphine, only one out of ten subjects had nausea lasting 2 hours. Nausea and vomiting in the postoperative setting are antagonized by IV naloxone, without diminishing analgesia, at titrated doses of up to 5 μg/kg/h (211). Fortunately, the incidence of nausea and vomiting seems to be much less with repeated epidural dosing and is low in patients with cancer or nonmalignant pain who require long-term spinal opioid therapy (191,192,196,212).
Cardiovascular Function
Cardiovascular and pain regulatory systems are closely coupled. Activation or inhibition of one system produces changes in the other, often through overlapping anatomic and neurochemical pathways (213). Just as analgesia has multiple components, such as sensory processing or emotion, key aspects of global cardiovascular status—myocardial contractile state,
blood pressure, heart rate, and vascular resistance—although linked, are independently regulated. The bothersome side effect of dependent edema, sometimes observed during chronic spinal opioid therapy (particularly with morphine) likely is mediated through mechanisms unrelated to the just-mentioned parameters. It has been hypothesized, for example, that IT opioid migrating cephalad may stimulate the release of vasopressin (214). Cardiovascular effects of opioids differ between basal, pain-free subjects and those studied perioperatively or while in pain. Also, central injections of receptor-selective opioids in animals may elicit different, even opposite effects upon heart rate and blood pressure if given into nearby sites, or at the same site in low versus high doses. One explanation for dual excitatory and inhibitory effects parallels the explanation for biphasic opioid ventilatory effects, namely, that high-affinity μ1 receptors mediate excitatory effects and low-affinity μ2 receptors mediate inhibitory effects (213). Analgesic doses of spinal morphine do not change blood pressure or heart rate in animals and in humans studied under basal conditions in the awake or the anesthetized state (215,216). In halothane-anesthetized or unanesthetized dogs, IT morphine or fentanyl analogues do not change cardiac output or peripheral resistance (127,215). Dose-dependent reductions in heart rate were seen in awake dogs after either epidural or IT administration of boluses of sufentanil, alfentanil, or morphine, with tolerance to these effects during repetitive epidural or IT dosing (51). In resting humans, changes in skin temperature, blood pressure, and heart rate are absent with spinal opioids. Both sudomotor and vasomotor activity (e.g., responses to cold pressor testing or Valsalva maneuver) remain intact (39,173,192,210). The latter is important because of its homeostatic role during upright posture or blood loss.
blood pressure, heart rate, and vascular resistance—although linked, are independently regulated. The bothersome side effect of dependent edema, sometimes observed during chronic spinal opioid therapy (particularly with morphine) likely is mediated through mechanisms unrelated to the just-mentioned parameters. It has been hypothesized, for example, that IT opioid migrating cephalad may stimulate the release of vasopressin (214). Cardiovascular effects of opioids differ between basal, pain-free subjects and those studied perioperatively or while in pain. Also, central injections of receptor-selective opioids in animals may elicit different, even opposite effects upon heart rate and blood pressure if given into nearby sites, or at the same site in low versus high doses. One explanation for dual excitatory and inhibitory effects parallels the explanation for biphasic opioid ventilatory effects, namely, that high-affinity μ1 receptors mediate excitatory effects and low-affinity μ2 receptors mediate inhibitory effects (213). Analgesic doses of spinal morphine do not change blood pressure or heart rate in animals and in humans studied under basal conditions in the awake or the anesthetized state (215,216). In halothane-anesthetized or unanesthetized dogs, IT morphine or fentanyl analogues do not change cardiac output or peripheral resistance (127,215). Dose-dependent reductions in heart rate were seen in awake dogs after either epidural or IT administration of boluses of sufentanil, alfentanil, or morphine, with tolerance to these effects during repetitive epidural or IT dosing (51). In resting humans, changes in skin temperature, blood pressure, and heart rate are absent with spinal opioids. Both sudomotor and vasomotor activity (e.g., responses to cold pressor testing or Valsalva maneuver) remain intact (39,173,192,210). The latter is important because of its homeostatic role during upright posture or blood loss.
During noxious stimulation in awake or anesthetized animals and humans, parasympathetic outflow decreases and sympathetic activity increases, as do levels of circulating catecholamines (58,183,217) (see Chapters 6 and 22). Heart rate, blood pressure, myocardial oxygen consumption, systemic vascular resistance, and ventricular vulnerability to fibrillation all increase in this situation. Opioid analgesia by spinal or systemic routes decreases all these parameters (218). One study in dogs reported an increase in cardiac vagal activity after thoracic epidural morphine (219), consistent with previous findings that systemic opioids stimulate both the central baroceptor reflex and vagal afferent activity. Clinical effects of spinal analgesia upon myocardial function have been most evident when local anesthetics, alone or supplemented with opioids, are administered via the thoracic epidural space (220). The greater effect of spinal local anesthetics than opioids upon cardiovascular function during pain or stress probably reflects the more complete sympathetic ablation possible with the former class of agents (221). However, in both experimental and clinical studies, spinal opioid analgesia has not yet been clearly proven to benefit cardiovascular morbidity and mortality beyond what might be attributed to effective analgesia alone (222). Attempts to draw conclusions from published clinical series have been impeded by the small numbers of patients studied, and the likelihood that advances in perioperative monitoring and treatment of myocardial ischemia have advanced sufficiently that it may be necessary to study thousands of patients in order to demonstrate further decrements in cardiovascular complications (222,223). Thus, identification of the impact, if any, of spinal opioid analgesia upon such complications may require pooling of results from studies of high-risk patients undergoing highly invasive procedures—a synthesis that is not yet possible.
Bladder Function
Spinal morphine in humans produces a naloxone-reversible inhibition of the volume-evoked micturition reflex (190) to a greater degree than is seen after systemic administration of equianalgesic doses. This effect seems to be most marked in young males, but demographics are unreliable because many patients treated acutely with spinal opioids are high-risk older patients undergoing major operations, after which they are routinely catheterized (214). It is not yet proven if the incidence varies with the opioid used, the duration of treatment, or between males and females. That these factors may be important is suggested by a series of 40,000 injections of epidural meperidine 50 mg for pain after cesarean section in which catheterization of the bladder was not necessary (208). In this large series, patients routinely had bladder catheterization intraoperatively and for the first 12 hours postoperatively and then did not require recatheterization. The apparently prophylactic effect of this approach is of interest, since another study reported a low incidence of urinary retention using this method compared with a high incidence with “in, out” catheterization intraoperatively followed by epidural morphine (224).
Urinary retention is a more frequently reported complication (37,225,226,227,228) of spinal opioids administered to volunteer subjects than spinal opioids administered to patients. Current evidence from volunteer studies indicates that urodynamic effects of epidural morphine are not dose-related within a range of clinically relevant doses but they are reversed by naloxone (225). In a dose–response study of epidural morphine for postoperative pain relief, Martin and associates found that the incidence of urinary retention was the same for doses of morphine of 0.5, 1.0, 2.0, 4.0, and 8.0 mg (185). This was consistent with a urodynamic study in 30 volunteers in whom increased bladder capacity and relaxation of detrusor muscle was similar for epidural doses of morphine of 2, 4, and 10 mg (225). In a study of postoperative patients using CO2 cystometry, a great variation in bladder response to epidural morphine was found on the day after surgery. Intravenous naloxone reversed bladder effects in those patients who developed urinary retention (229). Naloxone infusion of 5 μg/kg/hr in postoperative patients in one study reversed urinary retention with only minimal effects on analgesia (230), but others have reported that naloxone doses large enough to consistently reverse urodynamic changes after epidural morphine (e.g., 10 μg/kg/hr) may interfere with analgesia. The precise mechanism of spinal opioid interference with bladder function has not been elucidated, although μ opioid receptors are clearly implicated. Anecdotal reports suggest that the incidence of bladder dysfunction may be less with spinal fentanyl (231), meperidine (208,232), and methadone (233) than with morphine but conclusive comparative studies are not available.
Neurotoxic Potential, Inflammatory Mass Formation, and Pruritus
The neurotoxic potential of spinally administered opioids has been evaluated extensively in both preclinical and clinical settings (234). High doses of IT morphine in rats may produce two alarming syndromes: (a) convulsive seizures of the hind limbs and hyperreflexia in response to cutaneous stimuli, and (b) intense motor rigidity. Neither of these are antagonized by naloxone, implying mediation by nonopioids such as the algesic metabolite morphine-3-glucuronide or the inhibitory neurotransmitter glycine. This cautionary note is not paralleled by
clinical reports: dysesthesia is rare (235) and not clearly distinguishable from pruritus. Similar contrast exists between sobering preclinical evidence (including histopathology) of neurotoxicity and motor paralysis after IT administration of peptides including somatostatin, dynorphin, or δ-selective enkephalin analogs in the rat or dog, and the rarity of corresponding symptoms in clinical trials of these agents. Explanations for this disparity include species differences as well as inclusion criteria in clinical trials that select patients in whom motor effects are undetected because they are already bedridden or inactive.
clinical reports: dysesthesia is rare (235) and not clearly distinguishable from pruritus. Similar contrast exists between sobering preclinical evidence (including histopathology) of neurotoxicity and motor paralysis after IT administration of peptides including somatostatin, dynorphin, or δ-selective enkephalin analogs in the rat or dog, and the rarity of corresponding symptoms in clinical trials of these agents. Explanations for this disparity include species differences as well as inclusion criteria in clinical trials that select patients in whom motor effects are undetected because they are already bedridden or inactive.
Apart from the potential neurotoxicity of spinally administered compounds themselves, other factors relevant to the safety of spinal opioid administration are the compatibility of solutions of such agents with CSF and neural tissue, and inflammatory reactions provoked by intraspinal catheters. Solutions of opioids used in spinal injections (morphine, methadone, meperidine, fentanyl, alfentanil, lofentanil, and buprenorphine) and local anesthetics in normal saline have pH values that range from 4.52 to 6.85. When mixed with CSF, all lowered the pH of the CSF by 0.3 or less, but etidocaine lowered pH by 0.82, with clouding of CSF (236). High concentrations of local anesthetics alone, such as 5% lidocaine given intrathecally, are recognized in animal models and clinical reports to have the potential for irreversible neurotoxicity. In fact, clinical adverse events such as cauda equine syndrome led to the withdrawal of spinal microcatheters after otherwise promising use (237). In retrospect, those neurotoxic events were very likely due to the catheters’ use to deliver 5% lidocaine rather than the catheters per se. Currently, cautious re-exploration of this potentially valuable technique for perioperative anesthesia and analgesia is proceeding, with reliance upon less problematic solutions such as isobaric (0.5%) bupivacaine (238). Microcatheters are also useful for trials of subacute IT administration of opioids to predict responses to chronic delivery via implanted pump.
Inflammatory mass formation is a problem of increasing concern. Histologic examination of spinal cords of cancer patients who had epidural administration of bupivacaine-morphine mixtures for 3 weeks (239) or morphine for up to 6 months (38) revealed no abnormality attributable to morphine. Two of seven patients had posterior column degeneration consistent with myelopathy of malignancy (240). Animal studies of the potential damage to the spinal cord of epidural catheters and repeated injection of opioids have revealed histologic changes in many but not all models. Yaksh reported that 14 macaque monkeys with epidural catheters in situ for 4 to 16 months and receiving 15 to 122 injections of morphine had no abnormal neurologic signs. Three of the monkeys were sacrificed for autopsy after 6, 8, and 9 months and after 44, 68, and 72 IT injections of a variety of opioids and peptides. No histologic evidence of cord pathology was found (128,241). Similar findings during chronic epidural or IT infusions of sufentanil, alfentanil, morphine, or saline in the dog were later reported by Yaksh and colleagues. In that study, every animal had fibrosis around its infusion catheter whether IT or epidural, and many had histologic evidence of acute and chronic inflammation in the epidural space. Animals with IT catheters, but not epidural catheters, also had acute and chronic inflammation in the meninges. The incidences of all these histologic changes were independent of the dose of opioid used, and were equally likely to occur in animals infused with saline alone. Abouleish and co-workers (239) found no immediate or chronic (42 days) changes in spinal cord histology attributable to a single IT injection of a large dose of morphine (0.07 mg/kg). Coombs and co-workers, however, reported extensive pericatheter reactions in sheep after chronic epidural infusions of high concentrations of hydromorphone and morphine, but not saline. These reactions were sufficient to produce spinal cord compression, parenchymal damage, and hind limb weakness.
In cats with intrathecally implanted catheters receiving only IT saline and killed 19 to 21 days after implantation, an inflammatory response developed, with a thin fibrotic sleeve surrounding the catheter with no obstruction to the tip. Where the catheter lay in contact with the spinal cord, mild deformation and local demyelination were present. Animals that received the ED100 for alfentanil or sufentanil daily for 5 days showed spinal cord pathology indistinguishable from those receiving saline. Animals receiving ten times the ED100 of either drug showed results similar to controls (127). Chronically implanted epidural catheters in rats resulted in the rapid development of a fibrotic reaction in 36 of 43 rats after only 1 day. After 10 days of catheterization, a thick fibrotic reaction obstructing the catheter tip was observed in 31 of 33 rats. Injection of methylene blue showed no spread into the epidural space; the dye filled the lumen of the catheter and then the sheath, making a blue spot on the skin if the injection was continued. A mild deformation of the dura was observed in all animals (242). Perhaps the most detailed animal studies of catheter-tip inflammatory masses were reported in adjoining papers by Yaksh’s group in dogs (243) and Hassenbusch’s group in sheep (244), together with an editorial by Follett (245). Figure 40-5 (kindly provided by Professor Yaksh) depicts the rostrocaudal development of a typical inflammatory mass in a dog given an IT infusion of morphine (12.5 mg/mL at 40 μL/hr) for 28 days. No infectious process was detected.
Consideration of these animal studies, and surveillance studies based upon registries of patients who have received implanted spinal drug delivery systems, identify high local concentrations or cumulative doses of opioids, particularly morphine, as risk factors (246). In a “Dear Doctor” letter, however, the manufacturer cautioned that “no dose and/or concentration of morphine sulfate can be considered completely free of risk from inflammatory mass” (247). Intrathecal baclofen, particularly when compounded in nonstandard concentrations or together with opioids, has also been associated with granuloma formation (26,27). The occurrence of catheter tip granuloma formation is estimated at about 1% at 6 years (246), a figure that appears to be rising owing to closer surveillance. Among the tens of thousands of patients worldwide with implanted spinal drug delivery devices, hundreds are known to have developed this problem (245) and more are anticipated, as the average duration of IT infusion therapy increases. A consensus approach endorsed by many leaders in spinal infusion therapy (25) recommends decreasing the risk at the outset of therapy by placing the catheter tip below the conus medullaris and using the lowest doses/concentrations of opioid possible. Because IT clonidine (248,249) and bupivacaine appear much less likely to produce inflammatory masses, either or both of these agents may be coadministered from the outset of therapy with the objective of reducing opioid requirements. Baseline documentation of the patient’s neurologic history and physical examination, plus radiologic images of the catheter location, are useful for future comparison. Symptoms that raise the index of suspicion for fibroma formation include an apparent loss of analgesic effectiveness, increased pain intensity, lower extremity weakness, and other neurologic sensorimotor deficits such as bowel or bladder dysfunction (24,247). Magnetic resonance imaging [MRI] with gadolinium enhancement is preferred for diagnostic imaging, but computed tomography [CT] myelography may also be used. The neurologic deficits associated with inflammatory catheter tip masses appear due to a compressive mass effect, rather than the neurotoxic changes described earlier for spinal opioids per se. Treatment should consist of cessation of
the infusion (while providing systemic opioids to avert an abstinence syndrome) and expectant observation. Cases in whom this conservative approach does not result in improvement may be considered for surgical excision of the mass, catheter removal, and replacement of the catheter tip two or more levels above or below the mass.
the infusion (while providing systemic opioids to avert an abstinence syndrome) and expectant observation. Cases in whom this conservative approach does not result in improvement may be considered for surgical excision of the mass, catheter removal, and replacement of the catheter tip two or more levels above or below the mass.
Pruritus is common during spinal opioid therapy but incidence estimates vary widely as its presence (especially if mild) may only be revealed on direct questioning. It seems likely that generalized pruritus associated with spinal opioids is due to widespread alteration in sensory modulation, since it occurs when there is evidence of opioid migration over the entire spinal cord to the brain. Sensory modulatory mechanisms in the upper cervical spinal cord and trigeminal system may be involved, since the onset coincides with the spread of hypalgesia to this region (210). Facial pruritus, which often is reported, may be explained by rapid penetration of opioid to the superficially placed caudal portions of the trigeminal nucleus (137). Pruritus is not due to preservatives, since it occurs with preservative-free opioids, nor is it likely to be due to histamine release (250), since its onset is approximately 3 hours after epidural or spinal opioid administration (172,173,210). Further, it occurs with fentanyl, which does not cause histamine release, as well as with morphine, that does (251). Such benefits as clinicians have observed with antihistamines given for pruritus due to spinal opioid therapy may well derive from their nonspecific sedating actions.
Putative mechanisms of pruritus after spinal opioid therapy include some that involve μ opioid receptor activation, such as stimulation of neuronal Gs (as described earlier) or inhibition of inhibitory interneurons in spinal cord, or stimulation of an itch reflex localized in the trigeminal nucleus. Mechanisms dependent upon μ receptor activation underlie naloxone successful therapy of spinal opioid-induced pruritus (210,230) or other agents with μ-antagonist properties, such as butorphanol or nalbuphine. Indeed, in one obstetric series, the incidence of pruritus was minimal with epidural buprenorphine or butorphanol, but 50% to 60% with epidural morphine or fentanyl (185). In volunteer studies of epidural morphine, 10 mg, pruritus occurred in 100% of subjects in one study (37) and in three of four subjects in another study (227). In postoperative patients, pruritus was present in 28% of patients receiving epidural morphine 10 mg in one study (179) but in only 1% of patients in studies of morphine 5 mg (37) and 2 mg, respectively (166). Pruritus also has been reported with epidural meperidine, fentanyl, alfentanil, sufentanil, and diamorphine, but few comparative data are available. After meperidine 50 mg was given epidurally for postcesarean section pain, Brownridge reported that 50% of 2,000 patients admitted to pruritus (but only on direct questioning), yet only one patient found it troublesome (208). In contrast, in some series, epidural morphine resulted in pruritus in up to 70% of patients, but there was no relationship between incidence of pruritus and dose (250). However, the incidence of severe pruritus that troubles the patient appears to be close to 1% (37). Other putative mechanisms of spinal opioid–induced pruritus that are independent of the μ receptor include the formation of hyperalgesic metabolites such as morphine-3-glucuronide. This metabolite produces scratching behavior and hyperalgesia not reversed by naloxone after injection into monkeys. Further evidence that pruritus need not follow activation of spinal μ opioid receptors is the absence of pruritus after IT administration of β-endorphin (252,253). Hyperesthesia of unclear etiology but not reversible by naloxone, has been observed after high doses of spinal morphine given to patients with cancer pain (254,255). Fortunately, even high doses of sufentanil and alfentanil do not appear to be associated with this side-effect (255). It has been reported that prior or concomitant use of bupivacaine epidurally reduces the incidence of pruritus with epidural opioids (256). The results of one recent trial suggest that subhypnotic doses of propofol may be an attractive novel means of treating pruritus (257). Pruritus often subsides, as does bladder dysfunction, with continuing doses of opioid.
Endocrine Dysfunction—Hypogonadism
An extensive literature in opioid addicts, patients on methadone maintenance (261,262), and animals has linked opioid administration with impairment of endocrine function. Exogenous opioids mimic an endogenous negative feedback loop (259) in which β-endorphin derived from pituitary proopiomelanocortin inhibits the secretion of hypothalamic corticotrophin-releasing hormone (260,261). However, this phenomenon (the subject of rare case reports) appears to have minimal clinical importance. Yet endogenous opioids do mediate stress-induced hypogonadism through a presumably protective reflex that inhibits male and female reproductive function during acute and chronic illness (including depression) or stressors such as caloric siphoning due to intensive prolonged exercise (263). Therefore it is not surprising that chronic therapy with systemic (264) or spinal opioids (265,266,267,268) should give rise to secondary hypogonadism.
The most common endocrine issue related to systemic or central opioid use is hypoandrogenism, both in men and women (265,269). Female hypogonadism is equally likely with chronic opioid use but typically is manifest as loss of libido rather than poor sexual performance. Both male and female opioid-induced hypogonadism may present as accelerated osteoporosis (e.g., hip or vertebral fractures) (270). Estimates of the prevalence of opioid-induced diminished libido and sexual dysfunction vary widely due to different dose ranges employed in the clinical series, different underlying diagnoses and comorbid conditions, varying age ranges reported, and other factors. Patients who note newly decreased libido during chronic opioid therapy regardless of administration route should be evaluated as one would if opioids were not a possible etiologic factor. The clinician should inquire about symptoms frequently associated with hypogonadism: loss of libido; impotence; loss of gender role; depression, anxiety, fatigue; loss of muscle mass and strength; (in females) olio- or amenorrhea (271), galactorrhea; (possibly) worsening of pain; and/or accelerated osteoporosis. The contribution of causes associated with loss of libido and/or erectile dysfunction other than opiotherapy should be assessed, including family stress in chronic pain. Such causes include normal age-related decline, pain during intercourse (e.g., from lumbosacral spine pathology or peripheral nerve dysfunction such as ilioinguinal), weight gain, vascular dysfunction per se or related to other intercurrent conditions such as diabetes mellitus or liver disease, nonopioid medications, unrelieved pain, and psychological conditions per se or known to be more likely with chronic pain (anxiety, depression), including marital discord. Physical examination should assess for obesity, peripheral neuropathy, and in men (who are more likely to note symptoms) testicular atrophy, gynecomastia, and decreased beard. Point tenderness upon spine percussion may indicate compression fracture. Laboratory evaluation should include free and total testosterone, keeping in mind that there is a normal age-related decline in testosterone (272). A rule of thumb is that male hypogonadism is excluded if the total (bound + free) testosterone exceeds 300 ng/mL. Low or normal plasma levels of luteinizing hormones and follicle stimulating hormone
in the presence of subnormal testosterone levels confirm the diagnosis of secondary hypogonadism due to opioids. A bone densitometry test is useful to disclose bone mineral loss prior to vertebral or long bone fracture.
in the presence of subnormal testosterone levels confirm the diagnosis of secondary hypogonadism due to opioids. A bone densitometry test is useful to disclose bone mineral loss prior to vertebral or long bone fracture.
Therapy for opioid-induced hypoandrogenism may be as simple as rotation of the opioid to permit dose reduction (124). Should that not work, nor opioid doses lowered by the addition of nonopioid adjuvant medication(s), then blood levels of prostate-specific antigen (PSA) should be checked. If the PSA level is normal, testosterone replacement may be started using depot intramuscular injections, oral replacement, or transdermal gel. The goal is to have a total plasma testosterone level of 300–1,200 ng/mL. Monitoring of PSA levels, complete blood count (because of possible erythrocytosis), and lipid profile are recommended. If testosterone therapy is inadvisable, then sildenafil or a similar agent for erectile dysfunction may be used at normal doses, providing there are no contraindications to the latter (e.g., nitrate therapy).
Spinal Opioid Pharmacology
The term selective spinal analgesia was suggested by Cousins and associates (39) in 1979 (see later discussion) to emphasize the distinction between spinal opioid analgesia versus analgesia achieved by relatively nonselective blockade of axonal conduction by local anesthetics. From the prior section, it is clear that spinal opioids have many effects other than antinociception. However, animal and human studies have amply validated the concept of “selective spinal analgesia” mediated by multiple opioid and nonopioid spinal antinociceptive systems. Accumulating evidence points to the feasibility of “combination spinal analgesic chemotherapy” (32) to achieve the ideal of analgesia without unwanted side effects by coadministering two or more agents, such as opioids and local anesthetics, that act upon different antinociceptive targets. This section summarizes the preclinical and clinical data that model the pharmacokinetics and pharmacodynamics of IT and epidural opioids given as sole agents or in combination with other opioid and nonopioid compounds.
Physicochemical Properties
Local anesthetics (273) and nonpeptidic opioids have similar molecular weights and pKa values; partition coefficients for individual agents within both drug classes overlap but vary widely (Table 40-4).
Phenylpiperidine derivatives (meperidine, fentanyl, lofentanil) are highly lipid soluble and closest in structure to local anesthetics. In contrast, morphine has low lipid solubility. Morphine’s slow onset of action after epidural dosing coincides with delayed peak morphine concentrations in CSF (274,275), and its relative hydrophilicity results in slower efflux from spinal cord and CSF, and greater migration to the brain (135,276). Like lidocaine, meperidine has a rapid onset of analgesia after epidural dosing that coincides with an early peak drug concentration in CSF (39,192,275). High local concentrations of meperidine can produce peripheral nerve block (277), a property that is shared to an extent by other opioids, particularly nonalkaloids, along with many other classes of drugs used in anesthesia. Such high concentrations are unlikely to be infused spinally, and meperidine is the only opioid that has significant local anesthetic actions when applied to individual dorsal root axons or that is effective when used as a sole IT agent for surgery (278,279).
Table 40-4 Physicochemical properties of opioids and local anesthetics | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Onset of analgesia after spinal opioid administration is earlier with more lipid-soluble agents. Other factors such as molecular size and shape also contribute to a degree (280). Morphine, an extreme example of low lipid solubility and high water solubility, has the slowest onset of action. A somewhat quicker onset of clinical analgesia for hydromorphone than morphine after lumbar epidural administration despite comparable lipophilicity and similar blood and CSF pharmacokinetics (281) may reflect more rapid supraspinal action of the former compound (282). Many studies in animals (18,127,241) and humans (157,171,276,283) indicate the importance of lipid solubility for dural transfer (275,284).
Duration of analgesia is inversely related to lipid solubility but also influenced by the rate of dissociation from receptors and accumulation within epidural fat. In rats, the duration of analgesia for fentanyl analogues and morphine in hot plate and tail flick tests is dose-dependent (127). At doses producing an equal magnitude of peak analgesia, the duration of action was lofentanil >morphine >sufentanil >alfentanil ≥fentanyl.
Preclinical Pharmacokinetic Observations
Gustafsson and co-workers performed lumbar IT injections of [14C]-morphine and [3H]-meperidine in rats (285). Whole-body autoradiography revealed that 15 minutes after [14C]-morphine injection, the entire spinal cord and ventral parts
of the brain contained high levels of radioactivity. At 60 minutes, parts of the brain, including respiratory and vomiting centers and the trigeminal nucleus, contained radioactivity that remained detectable at 2 but not 4 hours after injection. Only the caudal part of the spinal cord contained radioactivity at 4 hours. Overall, the more lipophilic drug meperidine is rapidly taken up and eliminated from the spinal cord, whereas the hydrophilic drug morphine persists in the spinal cord for much longer. Also, morphine spreads rapidly into basal cisterns and later penetrates into brain.
of the brain contained high levels of radioactivity. At 60 minutes, parts of the brain, including respiratory and vomiting centers and the trigeminal nucleus, contained radioactivity that remained detectable at 2 but not 4 hours after injection. Only the caudal part of the spinal cord contained radioactivity at 4 hours. Overall, the more lipophilic drug meperidine is rapidly taken up and eliminated from the spinal cord, whereas the hydrophilic drug morphine persists in the spinal cord for much longer. Also, morphine spreads rapidly into basal cisterns and later penetrates into brain.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


