The most common inciting event in cardiogenic shock is an acute MI. Historically, once 40% of the myocardium has been irreversibly damaged, cardiogenic shock may result. From a mechanical perspective, decreased cardiac contractility diminishes both stroke volume (SV) and CO (see Table 44.2). These lead to increased ventricular filling pressures, cardiac chamber dilatation, and ultimately univentricular or biventricular failure with resultant systemic hypotension; this further reduces myocardial perfusion and exacerbates ongoing ischemia. The end result is a vicious cycle with severe cardiovascular decompensation. Similar to hypovolemic shock, a significant systemic inflammatory response has been implicated in the pathophysiology of cardiogenic shock.
Obstructive Shock
In obstructive shock, external forces compress the thin-walled chambers of the heart, the great vessels, or any combination thereof. These forces impair either the diastolic filling or the systolic contraction of the heart (see Table 44.1). Large obstructive intrathoracic tumors, tension pneumothoraces, pericardial tamponade, and constrictive pericarditis limit ventricular filling, while pulmonary emboli (PE) and aortic dissection impede cardiac contractility.
The hemodynamic parameters witnessed in obstructive shock include increases in central venous pressure (CVP) and SVR, and decreases in CO and mixed venous oxygen saturation (SvO2) (see Table 44.2). The PCWP and other hemodynamic indices are dependent on the obstructive cause. In pericardial tamponade, there is equalization of the right and left ventricular diastolic pressures, the CVP, and the PCWP (increased). However, following a massive PE, right ventricular failure leads to increased right heart pressures and a normal or decreased PCWP.
Distributive Shock
Distributive shock is characterized by a decrease in SVR. Septic shock is the most common form although, additionally, distributive shock includes the other oft-quoted classes of shock including anaphylactic, neurogenic, and adrenal shock (see Table 44.1). Physiologically, all forms of distributive shock exhibit a decreased SVR (see Table 44.2). Subsequently, these patients experience relative hypovolemia as evidenced by a decreased (or normal) CVP and PCWP. The CO is initially diminished; however, following appropriate volume loading, the CO increases.
CELLULAR ALTERATIONS
All forms of shock, especially hemorrhagic and septic, induce a host response that is characterized by local and systemic release of proinflammatory cytokines, arachidonic acid metabolites, and activation of complement factors, kinins, and coagulation as well as hormonal mediators. Clinically, this is the systemic inflammatory response syndrome (SIRS). Paralleling this response is an anti-inflammatory response referred to as the compensatory anti-inflammatory response syndrome (sometimes abbreviated CARS). An imbalance between these responses appears to be responsible for increased susceptibility to infection and organ dysfunction (25–29).
| TABLE 44.2 Shock Hemodynamic Parameters |
 |
| TABLE 44.3 Clinical Parameters of the Systemic Inflammatory Response Syndrome |
 |
Systemic Inflammatory Response Syndrome
In 1991, a consensus conference of the American College of Chest Physicians and the American Society of Critical Care Medicine defined SIRS as a generalized inflammatory response triggered by a variety of infectious and noninfectious events (30). They arbitrarily established clinical parameters through a consensus process; Table 44.3 summarizes the SIRS diagnostic criteria. At least two of the four criteria must be present to fulfill the diagnosis of SIRS. Note, this definition emphasizes the inflammatory process regardless of its etiology. Subsequent studies have validated these criteria as predictive of increased ICU mortality, and indicated that this risk increases concurrent with the number of criteria present. SIRS is characterized by the local and systemic production, and release, of multiple mediators, including proinflammatory cytokines, complement factors, proteins of the contact phase and coagulation system, acute phase proteins, neuroendocrine mediators, and an accumulation of immunocompetent cells at the local site of tissue damage (31).
Compensatory Anti-Inflammatory Response Syndrome
Shock stimulates not only the release of proinflammatory mediators, but also the parallel release of anti-inflammatory mediators (26). This compensatory anti-inflammatory response is present concurrently with SIRS (Fig. 44.1) (32). When these two opposing responses are appropriately balanced, the patient is able to effectively recover without incurring secondary injury from the autoimmune inflammatory response (25). However, overwhelming CARS appears responsible for postshock immunosuppression, which leads to increased susceptibility to infections and sepsis (26,31,33). With time, SIRS ceases to exist and CARS is the predominant force.

FIGURE 44.1 Postinjury multiple organ failure occurs as a result of a dysfunctional inflammatory response. SIRS, systemic inflammatory response syndrome; MOF, multiple organ failure; CARS, compensatory anti-inflammatory response syndrome.
Cytokine Response
Proinflammatory cytokines, tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β) are key to the resultant inflammation (34,35). Secondary proinflammatory cytokines are released in a subacute fashion and include IL-2, IL-6, IL-8, platelet-activating factor (PAF), interferon-γ (IFN-γ), endothelin-1, leukotrienes, thromboxanes, prostaglandins, and the complement cascade (34,36).
IL-6 also acts as an immunoregulatory cytokine by stimulating the release of anti-inflammatory mediators such as IL-1 receptor antagonists and TNF receptors, which bind circulating proinflammatory cytokines (35). IL-6 also triggers the release of prostaglandin E2 (PGE2) from macrophages (35); PGE2 is potentially the most potent endogenous immunosuppressant (35). Not only does it suppress T-cell and macrophage responsiveness, but it also induces the release of IL-10, a potent anti-inflammatory cytokine that deactivates monocytes (35). A listing of pro- and anti-inflammatory mediators may be found in Tables 44.4 and 44.5.
Cell-Mediated Response
Shock alters the ability of splenic, peritoneal, and alveolar macrophages to release IL-1, IL-6, and TNF-α, leading to decreased levels of these proinflammatory cytokines (35). Kupffer cells, however, have an enhanced capacity for production of proinflammatory cytokines. Cell-mediated immunity requires not only functional macrophage and T cells, but also intact macrophage–T-cell interaction (35). Following injury, human leukocyte antigen (HLA-DR) receptor expression is decreased, leading to a loss of antigen-presenting capacity and decreased TNF-α production. PGE2, IL-10, and TGF-β all contribute to this “immunoparalysis” (25,35).
T-helper cells differentiate into either TH1 or TH2 lymphocytes; TH1 cells promote the proinflammatory cascade through the release of IL-2, IFN-γ, and TNF-β, while TH2 cells produce anti-inflammatory mediators (25,35). Monocytes/macrophages, through the release of IL-12, stimulate the differentiation of T-helper cells into TH1 cells. Because IL-12 production is depressed following trauma, there is a shift toward TH2, which has been associated with an adverse clinical outcome (25,35).
| TABLE 44.4 Proinflammatory Mediators | |
 | |
| TABLE 44.5 Anti-Inflammatory Mediators | |
 | |
Adherence of the leukocyte to endothelial cells is mediated through the upregulation of adhesion molecules. Selectins such as leukocyte adhesion molecule-1 (LAM-1), endothelial leukocyte adhesion molecule-1 (ELAM-1), and P-selectin are responsible for polymorphonuclear leukocytes (PMNLs) “rolling” (25,37). Upregulation of integrins such as the CD11/18 complexes or intercellular adhesion molecule-1 (ICAM-1) is responsible for PMNL attachment to the endothelium (25). Migration, accumulation, and activation of the PMNLs are mediated by chemoattractants such as chemokines and complement anaphylotoxins (25). Colony-stimulating factors (CSFs) likewise stimulate monocyte- or granulocyto-poiesis and reduce apoptosis of PMNLs during SIRS. Neutrophil apoptosis is further reduced by other proinflammatory mediators, thus resulting in PMNL accumulation at the site of local tissue destruction (25).
Leukocyte Recruitment
Proinflammatory cytokines enhance PMNL recruitment, phagocytic activity, and the release of proteases and oxygen-free radicals by PMNLs. This recruitment of leukocytes represents a key element for host defense following trauma, although it allows for the development of secondary tissue damage (38–41). Recruitment involves a complex cascade of events culminating in transmigration of the leukocyte, whereby the cell exerts its effects (42). The first step is capture and tethering, mediated via constitutively expressed leukocyte selectin denoted L selectin; L selectin functions by identifying glycoprotein ligands on leukocytes and those upregulated on cytokine-activated endothelium (42). Following capture and tethering, endothelial E selectin and P selectin assist in leukocyte rolling or slowing (37,43–48). P selectin is found in the membranes of endothelial storage granules, termed Weibel–Palade bodies (45). Following granule secretion, P selectin binds to carbohydrates presented by P selectin glycoprotein ligand (PSGL-1) on the leukocytes (25). In contrast, E selectin is not stored, yet it is synthesized de novo in the presence of inflammatory cytokines (43,44). These selectins cause the leukocytes to roll along the activated endothelium, whereby secondary capturing of leukocytes occurs via homotypic interactions. The third step in leukocyte recruitment is firm adhesion, which is mediated by membrane-expressed β1– and β2-integrins (49–51). The integrins bind to ICAM, resulting in cell–cell interactions and ultimately signal transduction. This step is critical to the formation of stable shear-resistant adhesion, which stabilizes the leukocyte for transmigration (49–51).
Transmigration is the final step in leukocyte recruitment following the formation of bonds between the aforementioned integrins and immunoglobulin (Ig)-superfamily members (42). The arrested leukocytes cross the endothelial layer via bicellular and tricellular endothelial junctions in a process coined diapedesis (52). This is mediated by platelet endothelial cell adhesion molecules (PECAMs), proteins expressed on both the leukocytes and intercellular junctions of endothelial cells (42).
Proteases and Reactive Oxygen Species
Polymorphonuclear lymphocytes and macrophages are not only responsible for phagocytosis of microorganisms and cellular debris, but can also cause secondary tissue and organ damage through degranulation and release of extracellular proteases and formation of reactive oxygen species (ROS) or respiratory burst (25,39,40,41,53–55). Elastases and metalloproteinases, which degrade both structural and extracellular matrix proteins, are present in increased concentrations following trauma (25). Neutrophil elastases also induce the release of proinflammatory cytokines (25).
ROS are generated by membrane-associated nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase, which is activated by proinflammatory cytokines, arachidonic acid metabolites, complement factors, and bacterial products (56,57). Superoxide anions are reduced in the Haber–Weiss reaction to hydrogen peroxide by superoxide dismutase located in the cytosol, mitochondria, and cell membrane (25). Hydrochloric acid is formed from H2O2 by myeloperoxidase, while the Fenton reaction transforms H2O2 into hydroxyl ions (25). These free ROS cause lipid peroxidation, cell membrane disintegration, and DNA damage of endothelial and parenchymal cells (58–60). Oxygen radicals also induce PMNLs to release proteases and collagenase as well as inactivating protease inhibitors (61).
Reactive nitrogen species cause additional tissue damage following trauma (62). Nitric oxide (NO), which induces vasodilation, is generated from L-arginine by inducible nitric oxide synthase (iNOS) in PMNLs or vascular muscle cells and by endothelial NOS (eNOS) in endothelial cells (62). iNOS is stimulated by cytokines and toxins, whereas eNOS is stimulated by mechanical shearing forces (62,63). Damage by reactive oxygen and nitrogen species leads to generalized edema and the capillary leak syndrome (62).
Complement, Kinins, and Coagulation
The complement cascade, kallikrein–kinin system, and coagulation cascade are intimately involved in the immune response to shock. They are activated through proinflammatory mediators, endogenous endotoxins, and tissue damage. The classic pathway of complement is normally activated by antigen–antibody complexes (Ig M or G) or activated coagulation factor XII (FXIIa), while the alternative pathway is activated by bacterial products such as lipopolysaccharide (64–66). Complement activation following trauma is most likely from the release of proteolytic enzymes, disruption of the endothelial lining, and tissue ischemia; the degree of complement activation correlates with the severity of injury. The cleavage of C3 and C5 by their respective convertases results in the formation of opsonins, anaphylotoxins, and the membrane attack complex (MAC) (64–66). The opsonins C3b and C4b enhance phagocytosis of cell debris and bacteria by means of opsonization (64,65). The anaphylotoxins C3a and C5a support inflammation via the recruitment and activation of phagocytic cells (i.e., monocytes, polymorphonuclear cells, and macrophages), enhancement of the hepatic acute phase reaction, and release of vasoactive mediators (i.e., histamine) (52,65). They also enhance the adhesion of leukocytes to endothelial cells, which results in increased vascular permeability and edema. C5a induces apoptosis and cell lysis through the interaction of its receptor and the MAC (52,65,66); additionally, C3a and C5a activate reparative mechanisms (65). C1 inhibitor inactivates C1s and C1r, thereby regulating the classic complement pathway. However, during inflammation, serum levels of C1 inhibitor are decreased via its degradation by PMNL elastases (65).
The plasma kallikrein–kinin system is a contact system of plasma proteases related to the complement and coagulation cascades. It consists of the plasma proteins FXII, prekallikrein, kininogen, and factor XI (FXI) (67). Activation of FXII and prekallikrein is via contact, when endothelial damage occurs and exposes the basement membrane (67). Factor XII activation forms factor XIIa (FXIIa), which initiates the complement cascade through the classic pathway, whereas prekallikrein activation forms kallikrein, which stimulates fibrinolysis through the conversion of plasminogen to plasmin or the activation of urokinase-like plasminogen activator (uPA) (67); tissue plasminogen activator (tPA) functions as a cofactor. Additionally, kallikrein supports the conversion of kininogen to bradykinin (67). The formation of bradykinin also occurs through the activation of the tissue kallikrein–kinin system, most likely through organ damage, as the tissue kallikrein–kinin system is found in many organs and tissues including the pancreas, kidney, intestine, and salivary glands. The kinins are potent vasodilators, increase vascular permeability and inhibit the function of platelets (67).
The intrinsic coagulation cascade is linked to the contact activation system via the formation of factor IXa (FIXa) from factor XIa (FXIa). Its formation leads to the consumption of FXII, prekallikrein, and FXI while plasma levels of enzyme–inhibitor complexes are increased (25). These include FXIIa-C1 inhibitor and kallikrein-C1 inhibitor. C1 inhibitor and α1-protease inhibitor are both inhibitors of the intrinsic coagulation pathway (68,69).
Although the intrinsic pathway provides a stimulus for activation of the coagulation cascade, the major activation following trauma is via the extrinsic pathway. Increased expression of tissue factor (TF) on endothelial cells and monocytes is induced by the proinflammatory cytokines TNF-α and IL-1β (69–71). The factor VII (FVII)–TF complex stimulates the formation of factor Xa (FXa) and ultimately thrombin (FIIa) (25). Thrombin-activated factor V (FV), factor VIII (FVIII), and FXI result in enhanced thrombin formation (25). Following cleavage of fibrinogen by thrombin, the fibrin monomers polymerize to form stable fibrin clots. The consumption of coagulation factors is controlled by the hepatocytic formation of antithrombin (AT) III (25). The thrombin–antithrombin complex inhibits thrombin, FIXa, FXa, FXIa, and FXIIa (72); other inhibitors include TF pathway inhibitor (TFPI) and activated protein C in combination with free protein S (72). Free protein S is decreased during inflammation due to its binding with the C4b-binding protein (68,72).
Disseminated intravascular coagulation (DIC) may occur following shock. After the initial phase, intra- and extravascular fibrin clots are observed. Hypoxia-induced cellular damage is the ultimate result of intravascular fibrin clots. Likewise, there is an increase in the interactions between endothelial cells and leukocytes (68–70,73). Clinically, coagulation factor consumption and platelet dysfunction are responsible for the diffuse hemorrhage (68,71). Consumption of coagulation factors is further enhanced via the proteolysis of fibrin clots to fibrin fragments (68,71). The consumption of coagulation factors is further enhanced through the proteolysis of fibrin clots to fibrin fragments by the protease plasmin (25,69,74).
Acute Phase Reaction
The acute phase reaction describes the early systemic response following shock and other insult states. During this phase, the biosynthetic profile of the liver is significantly altered. Under normal circumstances, the liver synthesizes a range of plasma proteins at steady-state concentrations. However, during the acute phase reaction, hepatocytes increase the synthesis of positive acute phase proteins (i.e., C-reactive protein [CRP], serum amyloid A [SAA], complement proteins, coagulation proteins, proteinase inhibitors, metal-binding proteins, and other proteins) essential to the inflammatory process at the expense of the negative acute phase proteins. The list of acute phase proteins, both positive and negative, is shown in Table 44.6 (75,76).
The acute phase response is initiated by hepatic Kupffer cells and the systemic release of proinflammatory cytokines IL-1, IL-6, IL-8, and TNF-α (77,78). The acute phase reaction typically lasts for 24 to 48 hours prior to its downregulation (35). IL-4, IL-10, glucocorticoids, and various other hormonal stimuli function to downregulate the proinflammatory mediators of the acute phase response (35); this modulation is critical. In instances of chronic or recurring inflammation, an aberrant acute phase response may result in exacerbated tissue damage (35).
The major acute phase proteins include CRP and SAA, the activities of which are both poorly understood (79,80). CRP was so named secondary to its ability to bind the C-polysaccharide of Pneumococcus. During inflammation CRP levels may increase by up to 1,000-fold over several hours depending on the insult and its severity (35). It acts as an opsonin for bacteria, parasites, and immune complexes; activates complement via the classic pathway; and binds chromatin (35). Binding chromatin may minimize autoimmune responses by disposing of nuclear antigens from sites of tissue debris (35). Clinically, CRP levels are relatively nonspecific and not predictive of posttraumatic complications. Despite this fact, serial measurements are helpful in trending a patient’s clinical course (35).
| TABLE 44.6 Acute Phase Proteins | |
 | |
SAA interacts with the third fraction of high-density lipoprotein (HDL3), thus becoming the dominant apolipoprotein during acute inflammation (81). This association enhances the binding of HDL3 to macrophages, which may engulf cholesterol and lipid debris. Excess cholesterol is then utilized in tissue repair or excreted (35). Additionally, SAA inhibits thrombin-induced platelet activation and the oxidative burst of neutrophils, potentially preventing oxidative tissue destruction (35).
DIAGNOSIS OF SHOCK
Early diagnosis of shock affords the patient the best possible outcome. The patient in overt shock with hypotension and tachycardia is relatively easy to diagnose. However, more often than not, shock presents in more insidious forms, whereby underrecognition and delay in treatment can lead to a poor outcome. Moreover, the concurrent presence of mixed shock states can confuse the picture. Diagnosis of shock relies on both basic history and physical examination skills, as well as more advanced technology available to the clinician.
Numerous clues in a patient’s history may help alert the physician to the possibility of impending shock. Large fluid losses via traumatic or gastrointestinal hemorrhage, third spacing from intra-abdominal surgery or pancreatitis, prolonged dehydration from vomiting or diarrhea, or insensible losses from burns may very easily tip the patient into hypovolemic shock. A history of infection, presence of indwelling catheters, or recent surgery may be implicated in septic shock. Neurogenic shock occurs almost exclusively after trauma, although limited forms are seen with spinal anesthesia. History of prolonged steroid use, particularly in the elderly, may indicate adrenal shock in the patient with hypotension postoperatively. Exposures to drugs, transfusions, or other allergens should be sought to rule out anaphylactic shock. Recent MI or cardiac intervention can lead to pump failure and cardiogenic shock. A detailed history is especially important for obstructive forms of shock, in which any intervention involving the chest can lead to either immediate or delayed compromise via cardiac tamponade or tension pneumothorax. Likewise, a history of deep venous thrombosis (DVT) or risk factors for thrombosis should alert the physician to the possibility of acute massive PE in the hypotensive patient.
Physical examination can provide more clues than just basic blood pressure measurements. As noted previously, hypotension alone is neither exclusive to shock nor absolute for a diagnosis, and therefore is only a small component of the physical examination. Certain findings may vary based on the type and timing of shock. The end result of any form of shock, however, is diminished end-organ perfusion. Therefore, any signs or symptoms of organ dysfunction should be considered as possible indicators of shock (Table 44.7). Often, the first sign of shock manifests as mental status changes, whether excitatory or somnolent in nature. The patient may appear diaphoretic and clammy in cardiogenic shock or warm and dry in early distributive shock. Heart rate may also be variable with tachycardia compensating for diminished CO in the patient with intact sympathetic drive. Vasoplegic shock, such as neurogenic or adrenal (or in the β-blocked patient), may not have the compensatory increase in heart rate normally seen, and may itself provide a clue as to the type of shock. Tachypnea is almost universally seen, as the body tries to buffer the lactate produced in a state of tissue hypoxia. The kidneys provide a sensitive measure of adequate end-organ perfusion, as manifested by low urinary output. Cardiogenic shock has its own specific physical findings including increased venous jugular distension, acute pulmonary edema, and new murmurs or dysrhythmias.
| TABLE 44.7 Clinical Recognition of Shock |
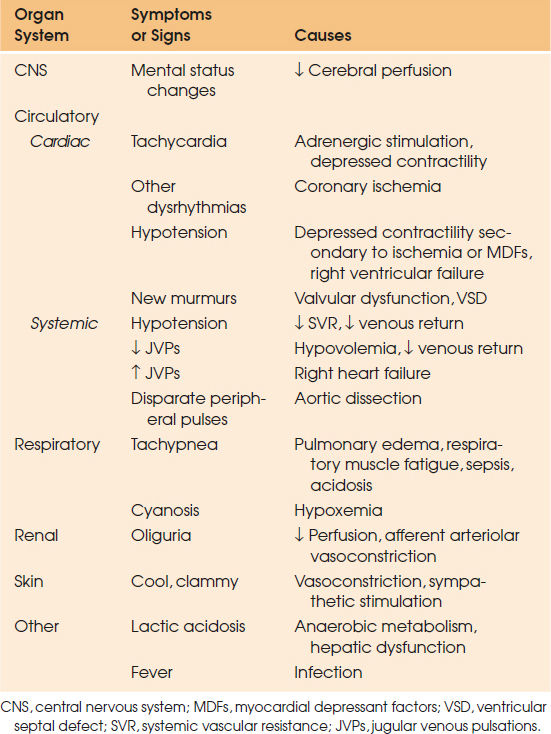 |
Various modalities for evaluating shock may be used either alone or in combination. Pooling data from multiple sources, however, is often required to get an adequate picture of shock resuscitation. Basic laboratory studies such as lactate level, base deficit, hemoglobin (Hgb), creatinine, and cortisol may help provide evidence of or reason for shock. Likewise, a more advanced evaluation of shock may include echocardiogram, CVP monitoring, tissue oxygenation and capnography, or advanced methods of determining CO. Advantages and disadvantages of these more advanced modalities will be discussed later within the context of shock monitoring.
MANAGEMENT OF SHOCK
Optimal management of shock depends first and foremost on early recognition of the syndrome, determination of its etiology, and correction of the underlying source while supporting the patient hemodynamically. Rapidity and adequacy of shock resolution will help prevent secondary reperfusion injury and prolonged morbidity. Variables of shock resuscitation must be frequently reassessed and therapy adjusted accordingly.
The underlying goal of shock management is to improve tissue oxygen perfusion. This may be accomplished by manipulating one or multiple physiologic parameters involved in oxygen delivery (DO2) and extraction. Forms of obstructive shock require the most prompt diagnosis, as continued mechanical impairment can be rapidly fatal. Adequate treatment of these etiologies can be just as rapid in the form of needle decompression for a tension pneumothorax or pericardiocentesis for cardiac tamponade. Management of distributive and hypovolemic forms of shock likewise involves source control early in the diagnosis. This may be in the form of hemorrhage control, removal of infected tissue (source control), or removal of an anaphylactic source. Once the inflammatory cascade has initiated, vasoactive medications are often used in addition to fluid provision to increase perfusion. Treatment of cardiogenic shock employs multimodality treatment including volume optimization, vasopressors, control of dysrhythmias, use of inotropes and the mechanical-assist devices, and early revascularization in primary myocardial ischemia (82).
Classically, all forms of shock are primarily treated with a combination of fluids and vasoactive agents. Deliberation is ongoing regarding the dosing and selection of these modalities for resuscitation, and will be examined here in greater detail.
Fluid Resuscitation
The initial treatment for all forms of shock is fluid administration. Provision of fluid helps restore perfusion and replace intravascular volume lost via hemorrhage, capillary leak, or redistribution. Intravenous fluid is readily available, inexpensive, easy to administer, and has low intrinsic morbidity. The etiology of shock and response to fluid will further dictate continued use of volume as primary therapy; however, all forms of shock potentially benefit from an initial fluid challenge (83). Deliberation should be given to the method of delivery, timing of administration, type of fluid, and volume of administration.
Route of Administration
The setting of shock dictates administration of fluid primarily via the intravenous route, which may be in the form of a peripheral or central venous catheter. Although the type of shock may guide the choice of catheter (i.e., an introducer catheter for a rapid infusion system or a triple lumen catheter for anticipated vasopressor therapy), the dictum of “two large-bore peripheral IVs” cannot be overstated (84). As per Poiseuille’s Law, width and length of the catheter dictates flow; therefore, a long, narrow, peripherally inserted central catheter will be of little utility when infusing a large bolus of fluid quickly. In the severely volume-depleted patient with collapsed veins, obtaining percutaneous venous access can prove difficult; saphenous vein cut-downs or interosseous access, particularly in the trauma or pediatric patient, can provide means of fluid administration in these extreme situations.
Timing and Volume of Administration
For forms of hypovolemic shock in particular, the concept of early restoration of intravascular volume to prevent circulatory collapse has long been recognized. In the hemorrhagic patient, volume resuscitation combined with source control may limit or prevent a state of irreversible shock, or the “lethal triad” of hypothermia, coagulopathy, and acidosis (85,86). The importance of the timing of volume loading is also paramount in all forms of shock, particularly in sepsis (87). Amplification of the previously described immune response can potentially be avoided if perfusion is restored early in the pathophysiologic process (88). Often the resuscitation process begins in the prehospital phase, with ambulance personnel administering crystalloid en route. Standard fluid boluses in the patient with shock typically amounts to 20 to 30 mL/kg at a time.
Overly aggressive fluid resuscitation both early and late in the course can be harmful in some circumstances. The concept of hypotensive resuscitation in the patient in whom mechanical control of bleeding has not been achieved—whether in traumatic injury, aortic aneurysm rupture, or gastrointestinal bleed—advocates for limited early aggressive fluid administration. Measures to raise blood pressure, particularly with fluid administration, may be counterproductive in this setting. In the penetrating thoracic trauma patient, early administration of large volumes of crystalloid has been shown to increase bleeding and subsequent mortality. Pushing fluid and intravascular volume beyond the initial phases of ischemia may propagate reperfusion injury and can be detrimental to further recovery. Restrictive fluid therapies for resuscitation have emerged in an effort to reduce the cardiac, wound healing, and pulmonary complications associated with large crystalloid infusions. Once patients have been stabilized, a more restrictive strategy of fluid administration can prevent subsequent morbidity.
Continued fluid administration beyond an initial bolus relies more on the patient’s pathology and response to treatment rather than on arbitrary numbers. Physical examination characteristics such as jugular venous distension, skin turgor, urine output, and basic vital signs may give clues to volume state, but are notoriously subject to interpretation. The examiner is often misled by the appearance of gross edema, insomuch as it has no bearing on effective extracellular fluid volume in the patient with capillary leak. New tools for approximating intravascular volume status are emerging to provide dynamic variables for the clinician to use when estimating appropriateness for further volume resuscitation. Measures such as stroke volume variation (SVV) and pulse pressure variation (PPV) can provide more accurate assessment of volume status and are replacing CVP and pulmonary artery pressures as primary tools in the ICU (89–92); these will be discussed further below.
Types of Fluid
Considerable debate abounds regarding the types of fluid to be administered for shock resuscitation. Often the determination to use crystalloid versus colloid depends on fluid availability, clinical scenario, and regional practice differences. The fact that there is so much debate over the preferred fluid type indicates the lack of conclusive evidence for the superiority of one fluid over another.
Crystalloids. Composed of varying amounts of electrolytes and sugar, crystalloids are inexpensive, require no special tubing or preparation, and pose little to no risk of adverse reaction. Crystalloids used in shock resuscitation are generally categorized as isotonic or hypertonic, describing the in vivo tonicity of the fluid. Typical isotonic crystalloids used are normal saline, lactated Ringer solution, or other commercially available combinations of electrolytes with sodium as the primary ion. Lacking protein components, the isotonic crystalloids readily distribute to the extracellular fluid compartment and will require larger volumes of infusion to maintain intravascular filling. Traditional philosophy dictates that a threefold volume of crystalloid to colloid is required for intravascular expansion; this ratio has recently been debated, however, and may actually be closer to a ratio of 1.5:1 when comparing crystalloid to 5% albumin (93).
Normal saline (0.9% saline solution) and lactated Ringer solution compromise the majority of isotonic crystalloid used for shock. Normal saline provides sodium with an equal amount of chloride for buffer; hypernatremia and hyperchloremic metabolic acidosis are therefore potential consequences of continued normal saline administration (94). Because of this, normal saline should be used for resuscitation typically only for head trauma patients, as hyponatremia can increase morbidity in this patient population.
While the tonicity is essentially the same, the electrolyte composition of lactated Ringer solution is physiologically closer to plasma, with inclusion of potassium and calcium, and reduction in chloride concentrations. Lactated Ringer is considered one of the “balanced” crystalloids as a different anion is used besides chloride to balance the cations in solution (95). A chloride restrictive strategy is associated with less acute kidney injury in the critical care setting, and therefore these types of balanced solutions are favorable.
Hypertonic Crystalloid. Combining the convenience of crystalloid with the tonicity of colloids, hypertonic saline (HTS) has emerged as an important tool in shock resuscitation. Hypertonicity of the sodium concentration promotes influx of fluid from the interstitial space. As such, HTS is advantageous for rapid, low-volume resuscitation for hypovolemic shock, particularly in situations where resources and space are limited, such as a combat setting. Hypertonic solutions also favorably impact immune modulatory function. Studies investigating hemorrhagic shock have found a decrease in neutrophil activation, and upregulation of anti-inflammatory cytokine production with use of HTS. Additional data suggest that HTS positively affects cardiac function in addition to volume expansion in septic shock (96,97).
While relatively safe compared to colloid infusion, the administration of high concentrations of sodium for volume resuscitation carries the concern for hypernatremia and hyperosmolarity. Compromise of renal function is likewise feared with high sodium and osmolar loads (98,99). Reports of hypokalemia, metabolic acidosis, and impaired platelet aggregation have also been documented with HTS use (100). Primary use of HTS is in the traumatic brain injury patient; small volumes should be used and electrolytes, creatinine, and serum osmolarity should be checked frequently to avoid the abovementioned complications.
Colloids. In reference to volume resuscitation, colloids generally consist of fluids that have a higher molecular weight based on composition consisting of protein or starches. These components increase the cost of colloids, make them susceptible to shortage, and mandate specialized tubing for delivery. The possibility of transfusion reaction is increased, as some of these compounds are derived from blood products. Likewise, allergic reactions can be noted with some of the synthetic formulations.
Conceptually, colloids more rapidly expand intravascular volume owing to their higher oncotic pressure. This effect may not necessarily persist beyond a few hours, especially in the critically ill patient in which capillary permeability is altered (101). In addition to more rapid volume expansion with less fluid infusion, this same increase in intravascular oncotic pressure has prompted the employment of colloids with the intent to reduce or prevent secondary edema; this effect has not been appreciated clinically, however. Studies reveal that edema formation is more dependent on fluid volume than on fluid type per se (102).
Albumin. First used for fluid resuscitation during World War II, albumin is a colloid derived from pooled human plasma and diluted with sodium. Preparations consist of 5% or 25% solution in quantities of 250 to 500 mL or 50 mL, respectively. As a blood product derivative, albumin is subject to disadvantages faced by other donated products—namely, periodic shortages, high acquisition costs, and refusal based on religious grounds. While transmission of viruses or other blood-borne diseases is theoretically a risk, only a few cases have been reported. Like any resuscitation fluid, patients are subject to sequelae of volume overload if infusion amounts are not monitored.
While indications for albumin use are broad, proven benefit to particular therapies is increasingly narrow. Numerous studies detailing poor prognosis with low serum albumin levels in critically ill patients prompted attempts to improve survival with intravenous supplementation (103–105). Compared with other colloid administration, albumin itself has no benefit in this patient population (106,107).
Albumin as a resuscitation fluid likewise has come under scrutiny. Previously, studies investigating albumin as a volume expander have been underpowered, prompting meta-analysis as the primary statistical measure of its worth. An initial Cochrane review comparing albumin to crystalloid examined 24 studies and found a 6% increase in absolute risk of death with albumin infusion (108). To confuse matters, subsequent meta-analysis of 55 studies showed no difference in mortality between albumin and crystalloid for resuscitation (109, 110). In 2004, the Saline versus Albumin Fluid Evaluation (SAFE) trial prospectively compared albumin to isotonic crystalloid for fluid resuscitation in a mixed ICU population (93); results showed no difference in morbidity or mortality overall with either fluid choice, although traumatic brain injury patients did show increased morbidity with albumin use. Subsequent multicenter studies demonstrated no difference in morbidity or mortality for albumin versus crystalloid in septic shock resuscitation.
Starches. Synthetic colloid polymers were developed for use in volume resuscitation in an attempt to retain the oncotic properties of albumin while decreasing cost and transfusion risk. Initial formulations of hydroxyethyl starch (HES) included high–molecular-weight moieties, accounting for an increased risk of coagulation and renal disturbances associated with their use (111–113). Lower–molecular-weight HES solutions were subsequently developed, with resultant fewer negative effects on bleeding, but concern for dose-dependent impaired renal function persisted (114).
While numerous studies have illustrated downregulation of proinflammatory cytokines with HES use, some of these results may be an effect of the efficiency of volume resuscitation, and not necessarily the fluid itself (115–117). Ongoing concerns about increase in renal failure and mortality in septic and mixed ICU populations prompted the U.S. Food and Drug Administration to add a warning regarding use of HES products in 2013. As such, HES use is not supported at this time.
Despite the theoretical advantage of colloids over crystalloids for shock resuscitation, there is no evidence from randomized controlled trials to demonstrate mortality difference. Studies demonstrating improved short-term gains with colloids use a heterogeneous population and/or fluid composition, making interpretation and application difficult. In larger studies, short-term physiologic gains made from colloid use do not translate in to longer-term improvement. As colloids are not associated with improvement in survival, and are considerably more expensive, it is hard to justify their use (118).
Special Fluid Considerations
Hemorrhagic Shock Resuscitation
Aggressive use of crystalloids during the Vietnam conflict resulted in improved mortality and reduction in renal failure, but also led to the emergence of acute lung injury and acute respiratory distress syndrome in the trauma population. Extensive use of crystalloids for trauma followed, with the popular concept of pushing fluids beyond supranormal resuscitation goals (119). Consequences of this large-volume approach are becoming more evident, with adverse cardiac, pulmonary, coagulation, and immunologic effects documented with massive crystalloid infusion (120).
With the recognition of the “blood lethal triad” of coagulopathy, acidosis, and hypothermia in the bleeding trauma patient, methods to physiologically break this cycle have come into play. Pushing crystalloids for shock resuscitation merely aggravates this pathway. Appropriate resuscitation in hemorrhagic shock includes measures such as damage control surgery and hemostatic resuscitation. The goals of damage control surgery are to stop ongoing hemorrhage and provide control of any visceral injury in a truncated manner such that the patient can return to the ICU for warming and resuscitation. Key to this approach is a massive transfusion strategy in which blood is provided in a balanced manner and in preference to crystalloid or other colloid during this time period. Since the patient bleeds whole blood, it makes physiologic sense to provide blood products in a manner that resembles that of whole blood. Major studies including PROMMTT and PROPPR have demonstrated survival benefits for hemorrhagic shock when providing platelets:FFP:RBC in as close to a 1:1:1 ratio as possible (121,122). The combination of early mechanical bleeding control with this hemostatic resuscitation is the current standard for hemorrhagic shock resuscitation.
Pharmacotherapy in Shock
Primary therapy for shock involves treating the cause and supplementation with fluids. When these modalities fail, vasopressors are typically employed as supplementation. Shock is not hypotension alone, however, and other agents can be used to compensate for the diminished tissue perfusion defined by this syndrome. Drugs used for shock will be examined here by the classifications of vasopressor, inotrope, and miscellaneous, although these categories may overlap to a degree.
Vasopressors
Vasopressors are generally given after an initial fluid bolus has failed or had marginal effect. Within the context of avoiding the consequences of excessive fluid administration, vasopressors may help limit volumes of fluid given; however, peripheral and end-organ vasoconstriction have their own adverse effects. Striking the balance between volume and vasopressors in the context of timing and type of shock is therefore a key component to resuscitation. With early recognition of shock, vasopressors can often be avoided by restoration of volume (123).
End-organ arterial autoregulation generally compensates for decreased MAP within a certain range; however, local vasoconstriction and vasodilatation may be unable to overcome extremes of perfusion. Administering catecholamine vasopressors may help improve MAP and therefore improve tissue perfusion by redistributing CO. The venous compartment also benefits from vasopressor therapy by decreasing compliance and therefore improving effective volume. Classifications of vasopressors consist of natural and synthetic versions of catecholamines (Table 44.8).
Norepinephrine. A naturally occurring vasopressor, norepinephrine is released by the postganglionic adrenergic nerves in response to stress. It has potent α-adrenergic effects, with less potent β1-stimulation. The α-adrenergic effects lead to increased systolic and diastolic blood pressure, with the addition of increased venous return via decreasing venous capacitance; this subsequently leads to increased cardiac filling pressure. Effect on the coronary arterial flow is enhanced via the increase in diastolic blood pressure. The β-adrenergic effects lead to increased chronotropic function, although this is limited by the baroreflex of vasoconstriction, resulting in zero net change in heart rate. Enhanced inotrope stimulation and stroke volume are likewise negated by an increase in left ventricular afterload, leading to a limited increase in CO.
Historically, the exaggerated peripheral vasoconstrictive properties of the drug have promoted a level of distrust leading to the often quoted “leave ‘em dead.” These fears are largely unfounded at indicated dosing ranges, and use of the drug may actually enhance renal function (124). The drug is safe and easily titratable, and lacks the tachydysrhythmic properties of other frequently used agents for shock. Resurgence in the use of norepinephrine has occurred with the recognition of its beneficial properties, and is now recommended as the first-line vasopressor in the treatment of shock (125).
Epinephrine. Epinephrine is the major physiologic adrenergic hormone of the adrenal medulla and represents the maximum in catecholamine stimulation. The agent potently stimulates α1-receptors with resultant marked venous and arterial vasoconstriction. These changes may lead to detrimental effects on regional blood flow, particularly on mesenteric and renal vascular beds. β-Effects lead to increased heart rate and inotropism. Due to counter effects of β2-vasodilation, the diastolic blood pressure is only slightly affected, with a lesser degree of increase in MAP than seen with norepinephrine. Stimulation of β2-receptors and blunting of mast cell response also makes epinephrine highly effective for anaphylaxis. Epinephrine has dose-dependent effects, with very low doses stimulating primarily β-receptors. This property makes epinephrine attractive as a primary inotrope; however, the range of that particular low dose varies with each patient and titration may prove dangerous. Epinephrine is advocated as a secondary pressor for septic shock, and as the primary pressor for cardiac arrest resuscitation.
| TABLE 44.8 Sympathomimetic Drugs |
 |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


