KEY POINTS
Malaria should be excluded as a cause of fever in any febrile patient who has traveled recently to a malaria-endemic area, or who may have been infected by contaminated blood through transfusions, needlestick injury, or other methods of direct infection.
Immunity to malaria may be short lived, so that even those who have been brought up in malaria-endemic areas can develop severe disease when they return as travelers.
Severe malaria is a medical emergency and ideally should be managed by experienced personnel in an intensive care setting.
Although Plasmodium falciparum is the most important cause of serious malaria, P vivax and P knowlesi can also cause severe and fatal infections.
Intravenous artesunate should be given as soon as practicable and alternative or additional diagnoses should be excluded.
Complications of malaria such as hypoglycemia, lactic acidosis, seizures, organ failure, and secondary infections should be diagnosed early by regular monitoring and managed aggressively.
Malaria can be particularly severe in immunocompromised individuals including those who have been splenectomized.
THE BIOLOGY OF MALARIAL PARASITES
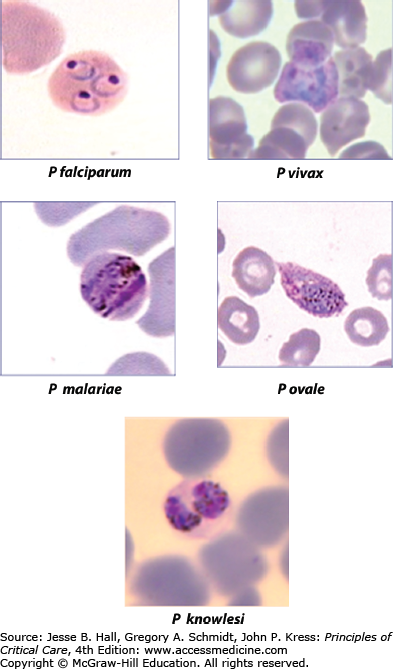
Since antiquity malaria has been a potent selective force on humanity’s history and its genes.1 Some Egyptian pharaohs carried malarial parasites2 and there are many descriptions of fevers in ancient cultures that are consistent with the diagnosis of malaria. Alphonse Laveran in Algiers in 1880 discovered that protozoan parasites cause malaria.3 Since then, four species of Plasmodium: P falciparum, P vivax, P malariae, and P ovale are established as causes of natural human malaria infections, with a fifth species, P knowlesi, usually infecting monkeys, emerging as an important human pathogen in some geographic areas4 (Fig. 78-1). All species of malarias are naturally transmitted to humans by female Anopheles mosquitoes when infected insects feed on blood. Annually, they account for an estimated 400 million episodes of malaria and 800,000 deaths.5 Deaths have been falling in recent years due to concerted efforts to control transmission of malaria, for example, by the use of insecticide treated bed nets, and the use of effective artemisinin combination therapies to treat clinical episodes. Most cases of severe malaria are attributed to infections with P falciparum although P vivax is increasingly recognized as causing severe morbidity in a minority of cases, as well as fatalities in some patients.6,7 P knowlesi can also cause severe disease and death.8 Mixed species infections are common in some areas.
When infected mosquitoes bite humans (Fig. 78-2), needle-shaped sporozoite stages of infection (approximately half a dozen) are injected and migrate within an hour to the liver to begin a clinically silent stage of intense multiplication within hepatocytes. One sporozoite can produce tens of thousands of progeny within 5 to 16 days, depending on species; the median time for P falciparum is 6 days. These progeny are then released into the bloodstream and multiply by asexual division in red cells until after several cycles of logarithmic growth sufficient parasites are generated to result in symptoms of disease. The numbers of parasites needed to produce symptoms in an individual vary with species of parasite, as well as being influenced by host factors such as prior exposure to malaria, which can produce variable immunity to disease. P vivax and P ovale can also generate curious dormant or hypnozoite stages of arrested development in liver cells. These hypnozoites can activate months after cure of the blood stages of infection by standard antimalarial treatments so that an additional course of an 8-aminoquinoline class of drug (primaquine) is needed to completely cure these species of infection.
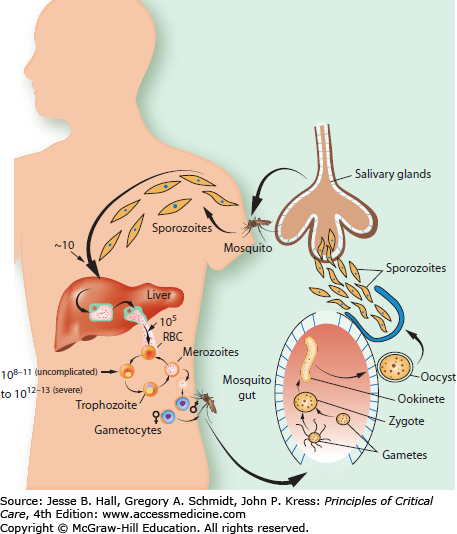
The time parasites take to replicate for one cycle in the blood depends on species: P knowlesi has a 24-hour cycle, and P malariae has a 72-hour cycle with the other species needing 48hours to feed, grow, multiply, and reinvade red cells. P falciparum can produce up to 36 merozoites or daughter cells, with other species producing sometimes as few as 4. Together with innate and acquired host defense mechanisms, the replicative cycle length and multiplicative capacity (number of progeny produced by one parasite each cycle) are important determinants of the total numbers of parasites in a human, and these numbers in turn are broadly related to risk of disease and death (discussed in greater detail below).9,10
EPIDEMIOLOGY
There are almost 100 tropical countries that are endemic for malaria with 67 of these implementing malaria control efforts of varying extent and 32 aiming to eliminate malaria. These concerted efforts to “shrink the malaria map” are having measurable and significant benefits in many areas, particularly those with moderate or low malaria endemicity.11 The degree of endemicity of infection refers to the risk to an individual of being exposed to bites of infected mosquitoes. This can range from more than one infected bite a day in some sub-Saharan countries, to less than one a year in many Asian countries. The age and duration of exposure to infected bites, together with other variables such as the presence of host resistance genes to malaria and nutritional status, act to influence the clinical features of disease (below). Even in tropical areas, Anopheline mosquitoes cannot transmit malaria when the environment is too cold (<16°C) or too hot (>33°C) or too dry, and altitudes >1500 m are also inhospitable for parasite propagation. This still leaves an estimated 1.4 billion individuals living with a stable risk of P falciparum malaria with about half in sub-Saharan Africa, half in Central and South East Asia, and 0.04 billion in the Americas. In Central Asia, many countries have transmission of both P falciparum and P vivax, whereas in Africa P vivax is found mainly in the Horn. P knowlesi is limited in its distribution to southeast Asia, where vectors (eg, of the A leucosphyrus group) can bite the natural hosts (long- and pig-tailed macaques) and also encounter humans. This primarily zoonotic means of transmission usually takes place when humans enter forested areas. P knowlesi has been mistaken for P malariae in these areas although the latter infection is more common in African countries.4 P ovale is found mainly in sub-Saharan Africa.
The force of selection imposed by P falciparum infections was suggested by JBS Haldane to be the reason why high heterozygote frequencies of blood disorders such as thalassemias were maintained in populations, because these hemoglobinopathies might confer a degree of protection against sequelae of infection. This hypothesis has stood the test of time and detailed investigations, and has been extended to include many other hemoglobinopathies (such as sickle cell disease12), red cell enzymopathies (such as G6PDH deficiency), red cell antigens (such as the Duffy blood group, which when absent makes red cells refractory to invasion by most P vivax, and P knowlesi13), as well as polymorphisms in host innate (such as cytokine and NO genes) and acquired defense genes.
PATHOPHYSIOLOGY
Theories on how infection with Plasmodium spp causes severe disease and death should accommodate the variable clinical spectra observed with different species of parasite, as well as age-related or geographic variations9,14,15 (see Table 78-1 for links between pathophysiology and disease). This spectrum extends from asymptomatic carriage of parasites (especially in older children and adults with acquired immunity in high transmission areas), to uncomplicated malaria that manifests as fever and other “flu-like” cytokine-mediated symptoms, to severe disease that involves one or more major organ systems and may cause lethal complications.
Manifestations and Complication of Severe Falciparum Malaria
| Manifestation or Complication | Pathophysiology | Treatment |
|---|---|---|
| Coma (Glasgow Coma Score <11; Blantyre Coma Score <3) and convulsions | Sequestration of parasitized red blood cells in the cerebral microcirculation and other factors | Hypoglycemia and other causes of meningo/encephalitis should be excluded. Good general intensive nursing care, including close observation of breathing, eye care, nasogastric tube, Foley catheter. If feasible: intubation to protect airway. Frequent monitoring of blood glucose. Treat convulsions (eg, lorazepam). Prophylactic anticonvulsive treatment is not recommended |
| Anemia (Hct <20%, in presence of parasitemia >100,000/μL) | Loss of parasitized red blood cells, increased splenic clearance of uninfected red blood cells (decreased red cell deformability, immunological factors?), dyserythropoiesis | General recommendation: transfusion if in distress, or Hct <20% (adults), or Hb <5 g/dL in children |
| Hyperparasitemia (>10% infected red blood cells) | Host immunological factors and parasite virulence factors (multiplication rate, red cell selectivity) | Promptly start parenteral antimalarial drugs in effective doses (artemisinins; if quinine: give loading dose). Exchange transfusion (?) |
| Hypoglycemia (blood glucose <40 mg/dL) | Increased use, decreased production, quinine related hyperinsulinism | Glucose 10%, 4 mg/kg bodyweight |
| Acute kidney injury | Acute tubular necrosis | Record fluid balance (Foley catheter) |
| Prerenal component (dehydration) | Check biochemistry (BUN, electrolytes), early start of renal replacement therapy (hemofiltration or hemodialysis preferred over peritoneal dialysis) | |
| Severe jaundice (bilirubin >3.0 mg/dL, with parasitemia >100,000/μL) | Mainly in adults; multifactorial | No specific treatment, monitor blood glucose |
| Fluid, electrolyte imbalances, metabolic acidosis (venous plasma bicarbonate <15 mmol/L or lactate >4 mmol/L) | Dehydration, SIADH (?), only minor increase in capillary permeability, compromised microcirculation by sequestration and other factors causing anaerobic glycolysis | Careful fluid resuscitation. Dialysis as treatment for severe acidosis has been advocated |
| Respiratory distress and pulmonary edema | Acidosis-related deep breathing. Pulmonary edema (ARDS) mainly in adults and pregnant women. Etiology unknown; cytokines mediated (?) | See also “acidosis”; ARDS: restricted fluid management, positive-pressure mechanical ventilation with PEEP etc. Do not allow “permissive hypercapnia” (cerebral edema) |
| Distinguish from pneumonia | ||
| Blackwater fever | Related to severe malaria, quinine use and G6PD deficiency | Transfusion if needed; bicarbonate administration (?) |
| Shock | Rare in malaria (NO binding by free hemoglobin?), consider concurrent bacteremia | Fluids, inotropic drugs (don’t use norepinephrine because of induction of lactic acidosis), antibiotics |
| Abnormal bleeding | Disseminated intravascular coagulation: consider concomitant bacterial infection | No specific treatment, packed red cell transfusion if indicated |
| Isolated thrombocytopenia (very common) |
Most research since the discovery of parasites has inevitably focused on P falciparum as this is more likely than other species to cause severe disease and death. P falciparum’s 48-hour red cell life cycle can be divided broadly into two halves. The first 24hours after invasion of red cells, parasites grow slowly and are relatively metabolically quiescent.16 These “ring” stages are most commonly visible in the peripheral blood so that regular examination of blood films not only aids in diagnosis but also allows monitoring of responses to treatment.10 As parasites mature during the second 24 hours of their red cell cycle, they increase rapidly in size and in their metabolic demands. There is enhanced glucose utilization (up to 100 times higher than that of uninfected cells) and concomitant lactate production by anaerobic glycolysis, increased degradation of hemoglobin and detoxification of heme iron by rendering it insoluble as crystalline hemozoin (parasite pigment), increased uptake of other synthetic precursors (for example of amino acids and choline) and remodeling of the red cell. These physiological changes are mediated by parasite-encoded transport proteins and are associated with modifications to the surface of the infected red cell that also increase its permeability.17 Such major changes to infected red cells might be expected to predispose them to increased clearance by the reticuloendothelial system, but additional adaptations described below can avoid this defense mechanism.
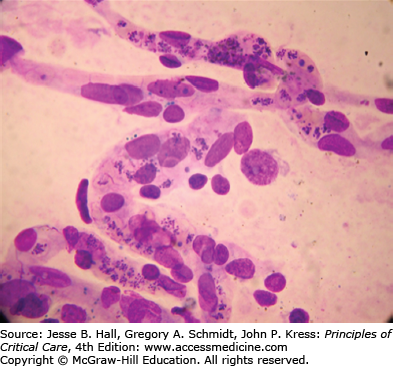
Parasites synthesize a family of proteins that are exported to the surface of red cells, where they mediate adhesion to endothelial cells, particularly in low flow regions such as capillaries and postcapillary venules. These proteins (collectively called Pfemp1) encoded by a var gene family (so-called, because they are antigenically highly variable, being exposed to selection by the immune system) can attach to several host receptors, including CD36, E-selectin, chondroitin-4-sulphate, and hyaluronic acid, on placental villi and members of the integrin family such as intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecular (VCAM).18 This phenomenon is called cytoadherence and is a key process in the pathogenicity pathway for severe disease. Cytoadherence causes sequestration of the infected erythrocytes in the microcirculation, obstructing flow19 (Fig. 78-3). Microcirculatory flow is further compromised by adherence of infected cells to each other mediated through their interaction with platelets (autoagglutination),20 or adherence to uninfected cells (rosetting). A reduction in deformability of both infected and uninfected erythrocytes further contributes to abnormal rheology and is a prognostic indicator of adverse outcomes.21 Sequestration of these late stages of parasitized cells prevents clearance from the circulation by conventional means.
P vivax preferentially invades and multiplies in reticulocytes, so parasitemia is limited by the number of circulating reticulocytes and young erythrocytes. P vivax can cause severe and sometimes fatal disease, although these complications are more likely to develop in those with chronic underlying morbidities such as renal impairment or diabetes. The degree to which P vivax also exhibits cytoadherence is not well studied, with some reports suggesting that P vivax can cytoadhere and form rosettes in ex vivo models.22,23 Unlike for many P falciparum infections, however, circulating parasites often include later stages of development suggesting that the numbers of adherent mature stages of P vivax are fewer.
P knowlesi is now an established cause of severe and fatal disease, and infections can be associated with very high peripheral parasitemias that are comparable in density to those seen in some P falciparum infections. The shorter (24hours) life cycle may also explain, at least in part, why later stages of development can also be seen on the peripheral blood film. In a single fatal case studied at postmortem, there was accumulation of parasitized erythrocytes in organ capillaries and venules, giving appearances similar to those of fatal cases of cerebral malaria caused by P falciparum, although coma was not a prominent clinical feature ante mortem.4,8 There is also some evidence that P knowlesi can cytoadhere to host ligands in ex vivo studies, providing further mechanistic insights into its pathophysiology.
P ovale and P malariae are also infections that have been identified in nonhuman primates, although they are not zoonoses, unlike P knowlesi. Their parasitemias are not usually very high, and these infections can persist for long periods of time. P malariae, for example, can persist for decades, and a chronic infection can cause an immune complex glomerulonephritis and nephrotic syndrome. They otherwise very rarely cause severe or fatal infections, unless they predispose to splenic rupture, which can take place with all species of malaria.
Cytoadherence of parasitized cells to blood vessels in different organs, and the release of host cytokines and other mediators when infected red cells rupture are important causes of disease and death due to malaria. Pigment produced by maturing parasites may contribute to both pathophysiological pathways by stimulating production of host cytokines as well as reducing red cell deformability and enhancing properties that favor cytoadherence. The relative contributions of cytoaherence and cytokines or other host responses to infection may vary with the genetic and immune status of the individual, as well as the species or strain of parasite. In one comparative study, P knowlesi patients had lower markers of macrophage activation such as MIP-1β and MCP-1 compared with P falciparum infections, while the anti-inflammatory cytokines IL-1ra and IL-10 were detected in all patients, including those with P vivax infection.24
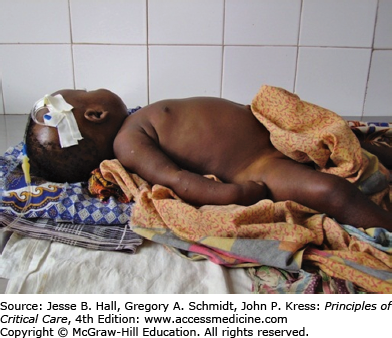
One of the most clinically discrete manifestations of severe disease caused by P falciparum infections is coma in the absence of other causes.25 Investigations over the past three decades have established that coma is associated with an increased density of parasitized erythrocytes in cerebral capillaries when compared with patients who died from severe malaria without coma.26-28 There is no large increase in blood-brain barrier permeability. Coma is also associated with increased anaerobic glycolysis,29 retinal hemorrhages and exudates and whitening of the retina,30 focal or generalized seizures, and clinical features of a metabolic encephalopathy such as dysconjugate gaze, bruxism, and abnormal extensor or flexor posturing31 (Fig. 78-4). Deep tendon, plantar, and abdominal reflexes are variably altered and may be increased or absent. These features are common to cerebral malaria in both African children and adults, but the syndromes in two age groups also differ in some aspects. CSF opening pressures are usually normal in adults with cerebral malaria, and usually elevated in children. Coma often recovers within a few hours in children and associated seizures (focal or generalized) are very common. Coma can take much longer (often 48-72hours) to reverse in adults. Approximately 10 % of African children are left with significant neurological sequelae, whereas adults usually make a full recovery.31 The radiological features of cerebral malaria may also differ between adults and children.
Both children and adults with malaria show many metabolic derangements associated with severe malaria. Hypoglycemia may occur in up to a quarter of children with cerebral malaria and is associated with other complications such as seizures and neurological sequelae. It is also associated with lactic acidosis and shares an underlying common mechanism of greatly increased anaerobic glycolysis from host tissues that have impaired microcirculation, as well as increased production of lactic acid due to anemia, seizures, fever, and decreased clearance of lactate by the liver.32 Acidosis may also interfere with compensatory shifts in the hemoglobin oxygen dissociation curve toward the right (Bohr shift) associated with anemia, by decreasing relatively the red cell synthesis of 2,3-diphosphoglycerate.33
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


