Epidemiology and Etiology
The term
juvenile rheumatoid arthritis (JRA) describes the condition of chronic arthritis of unknown cause in children <16 years and includes several subgroups
(Table 52.1). JRA is the most common chronic rheumatic disease in childhood with an estimated
incidence of 1:10,000 children per year and an estimated
prevalence of 1:1000. The gender ratio and age of onset differ for each subtype. JRA rarely presents before 6 months of age. Other chronic arthritides in children, not included under the term JRA, include psoriatic arthritis and spondyloarthropathies. The latter is also termed enthesitis-related arthritis (inflammation at the point of attachment of tendon to bone) or HLAB27-related arthritides. There is a new method of classification for all types of chronic arthritis in childhood of
unknown cause, under the umbrella term of juvenile idiopathic arthritis (JIA). This term is currently used more often by pediatric rheumatologists than JRA and has more scientific validity. However, since board questions relate to JRA, the term JRA was chosen for this chapter.
The causes of JRA are still unknown. However, genetic associations have been identified. Several human leukocyte antigen (HLA) genes have been associated with pauciarticular JRA and uveitis in young females with a positive antinuclear antibody (ANA). HLA-DR4 is associated with polyarticular JRA, particularly in rheumatoid factor (RF) and anticyclic citrullinated peptide (CCP) antibody positive polyarthritis found primarily in adolescent girls. Antibodies and cellular immune reactivity to type II collagen have also been found in children with JRA. Although several infectious agents have been reported as triggers, none have been consistently implicated in JRA. Polymorphisms of genes associated with macrophage as well as T- and B-cell activity are associated with an increased risk of developing JRA or a more severe clinical course.
Pathology
JRA is an autoimmune disorder that targets the synovial tissue of the joints. Examination of the synovial membrane shows villous hypertrophy and hyperplasia of the synovial lining that results in an increased secretion of joint fluid. Edema, hyperemia, vascular endothelial hyperplasia, and lymphocyte infiltration are seen. The inflammatory process, termed pannus, can eventually result in the erosion and destruction of articular cartilage and bone. Rheumatoid nodules are the consequence of small blood vessel vasculitis with a central area of necrosis and granulation. The rash of systemic-onset JRA is a result of neutrophil infiltration and perivasculitis. Inflammatory infiltrates may also be present in the liver and tissues such as pericardium, pleura, and peritoneum. The number of white blood cells, primarily polymorphonuclear leukocytes, in joint fluid varies from mild inflammation (5000/mm3) to purulent (>100,000/mm3). The severity of synovial leukocytosis does not correlate with the clinical course or outcome.
Clinical Presentation
JRA is classified into three subgroups according to the pattern of onset: pauciarticular, polyarticular, and systemic. The clinical manifestations vary among subgroups.
The arthritis of JRA is defined by the following:
Age at onset of less than 16 years
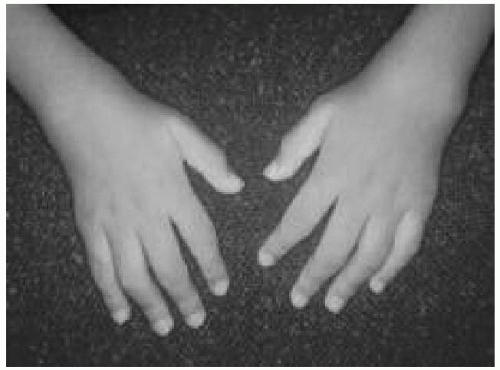
The presence of arthritis defined as articular swelling and effusion
(Fig. 52.1), or the presence of two of the following findings in joints (even in the absence of swelling): warmth, limitation of motion, and tenderness or pain on motion
Duration of arthritis of at least 6 weeks
Exclusion of other causes of arthritis
Other symptoms may include pain and morning stiffness, or stiffness after a nap or prolonged sitting. Children
may limp or refuse to use the affected limbs. Younger children often do not complain of pain and may present only with a limp or a regression in gross or fine motor skills. Anorexia, weight loss, and growth failure may also be seen, particularly in systemic or polyarticular-onset disease. Other extra-articular manifestations include:
Pauciarticular-Onset Juvenile Rheumatoid Arthritis
JRA with a pauciarticular onset accounts for approximately 40% to 50% of cases of JRA. Four or fewer joints are involved in the first 6 months of disease. The most common pattern is asymmetric involvement of the large joints of the lower extremities. About half of these children have monoarticular arthritis, mainly of the knee.
The onset of most cases of pauciarticular JRA is in early childhood. Children usually present between 1 and 4 years of age. Girls are more often affected than boys. The knees and ankles are typically involved. Hip involvement at the onset of pauciarticular JRA is very rare. Although the joints are swollen and warm, children often have very little pain and function well.
In nearly one third of cases, an asymptomatic chronic anterior uveitismay develop. Girls between 1 and 3 years of age with a positive ANA are at particularly high risk for developing uveitis. Chronic uveitis leads to significant visual impairment in up to 30% of patients; thus, prevention of permanent eye damage caused by uveitis is essential. Other common complications include the development of cataract, glaucoma, and band keratopathy. Therefore, a routine ophthalmologic slitlamp examination should be performed in all patients with JRA according to the schedule recommended by the American Academy of Pediatrics and the American College of Rheumatology. Young girls with pauciarticular JRA and positive ANA need an ophthalmologic examination every 3 to 4 months for the first 5 to 7 years of their disease.
Local complications of pauciarticular JRA include the development of flexion contractures, muscle atrophy, and leg length discrepancies. In younger children the affected limb is usually longer than the healthy limb, whereas in older children (>9 years) arthritis may lead to an early closing of the growth plate and a shorter limb. Thirty-three percent to 50% of children with pauciarticular JRA progress to develop a polyarticular course. Often, the outcome of uveitis and development of complications (glaucoma, cataract, visual defects) are the determinants of the long-term prognosis of pauciarticular JRA.
Polyarticular-Onset Juvenile Rheumatoid Arthritis
Approximately 35% of cases of JRA have a polyarticular onset. Five or more joints are involved in the first 6 months of disease. Characteristically, the arthritis is symmetric and affects both large and small joints. Hip, shoulder, and temporomandibular joint involvement are common. The cervical spine can also be involved and is associated with a risk for atlantoaxial subluxation. Many children in this subgroup have constitutional symptoms, including lowgrade fever, malaise, anemia of chronic disease, mild hepatosplenomegaly, and weight loss or growth delay.
There are two types of polyarticular-onset JRA. The more common type is RF negative polyarticular JRA. The onset is usually early in childhood (2-5 years old), and commonly affects large joints prior to small joints. Girls are affected more often than boys. Children respond well to treatment and have good functional outcomes, although the disease in most children persists to adulthood. Approximately 5% to 10% of these children develop chronic uveitis.
Only approximately 5% to 10% of children with polyarticular-onset JRA are positive for RF. Many of these children also have antibodies to CCP. The pattern of this disease resembles that of adult-onset rheumatoid arthritis. Girls, mainly adolescent, are affected more often than boys. Rheumatoid nodules or Felty syndrome (splenomegaly and leukopenia) may develop. The disease progresses rapidly, with joint erosions often evident within 6 to 12 months of onset of symptoms. Patients with HLA-DR4 typically have a worse prognosis. These patients should be treated aggressively early in the disease course.
Systemic-Onset Juvenile Rheumatoid Arthritis
Systemic-onset JRA is seen in 10% to 20% patients with JRA and can present at any age. This is the only type of JRA without a definite gender predilection. The arthritis of systemiconset JRA can be either pauciarticular or polyarticular,
affecting both large and small joints. Children with a polyarticular course often have severe disease with a poor outcome.
The arthritis is usually preceded by systemic manifestations for as long as even 6 months. Therefore, children often present with
fever of unknown origin and undergo extensive evaluations, especially for infection and malignancy (leukemia, neuroblastoma), before other diagnoses are excluded.
Fevers are intermittent, typically occurring in the evening or with two daily peaks. The
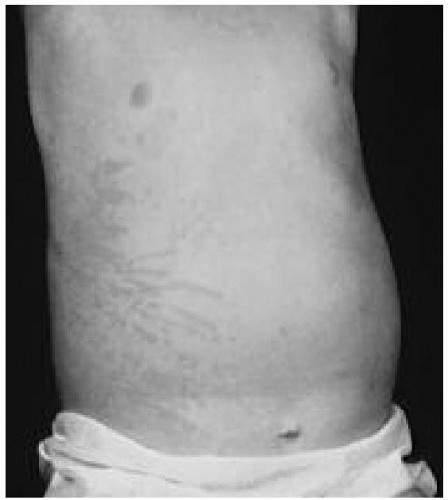
rash of systemic-onset JRA is transient, occurring more frequently during febrile periods, and is described as an erythematous or
salmonpink macular rash. This rash can often be elucidated by minor local trauma
(Koebner phenomena) or can be seen after a warm bath
(Fig. 52.2).
Lymphadenopathy and
hepatosplenomegaly are seen in more than two thirds of these children.
Pleuritis or
pericarditis is present in approximately 20% to 35% of patients. Rarely, pericarditis may be severe. There are no specific laboratory tests in systemiconset JRA. Laboratory results reflect an inflammatory state with a markedly increased sedimentation rate and C-reactive protein. Characteristic blood count findings include an elevated white blood cell count with a left shift, severe anemia, and thrombocytosis. Ferritin levels are often markedly increased. Serologic markers (ANA and RF) are usually absent. Uveitis is rarely found in this type of JRA.
Spondyloarthropathies (Enthesitis-Related Arthritis)
This class of arthritis usually presents in males (>80%) older than 8 years, with painful, asymmetric arthritis of the lower extremities (knees, ankles, tarsal joints, and hips) and often includes inflammation of the enthesis. Commonly affected enthesis are in the calcaneus, patella, and trochanter regions. An association with HLA-B27 is seen in most cases. Many of these children develop sacroiliitis or ankylosing spondylitis in adolescence or adulthood. A strong family history is often reported. Fifteen percent to 25% of these children develop acute uveitis, which unlike the uveitis of JRA, is associated with pain, erythema, and photophobia. Other forms of spondyloarthropathies include inflammatory bowel disease-related arthritis (both ulcerative colitis and Crohn disease) and postinfectious reactive arthritis (see subsequent section on Reactive Arthritis).
Psoriatic Arthritis
Fifteen percent to 20% of adults with psoriasis develop arthritis. Although uncommon, psoriatic arthritis is occasionally seen in children (approximately 5% of cases of chronic childhood arthritis). Psoriasis can occur simultaneously with arthritis, precede the development of arthritis by many years, or occur years after the beginning of arthritis. Several clues to the possibility of psoriatic arthritis in children include a family history of psoriasis in a first-degree relative, nail abnormalities (pitting, onycholysis), or tenosynovitis near small joints often in the pattern of dactylitis (sausage digit). Children with psoriatic arthritis can develop a pauciarticular, polyarticular, or spondyloarthropathy pattern of arthritis.
Diagnosis
The diagnosis of JRA is primarily based on the clinical findings and exclusion of other causes of arthritis. No specific diagnostic laboratory tests for JRA are available, although most patients with pauciarticular (50%-75%) and many patients with polyarticular JRA (40%-50%) have a positive ANA. The erythrocyte sedimentation rate and level of C- reactive protein may be elevated during active disease, but normal acute phase reactants do not rule out JRA. Approximately 5% to 10% of children with JRA are RF and anti-CCP antibody positive. Radiographs of the affected joints in the early stage of disease may show soft tissue swelling, regional osteoporosis, and demineralization. Later signs include bony overgrowth, joint space narrowing, erosions, subluxation, ankylosis, or joint destruction. In questionable cases, magnetic resonance imaging (MRI) (with gadolinium enhancement) may be helpful in the diagnosis of synovitis and the detection of joint damage even before conventional radiographs are done.
Treatment
The goals of therapy for JRA are to decrease the inflammatory process so as to minimize pain and joint damage, improve the function, and decrease the long-term disability. Nonsteroidal anti-inflammatory drugs are considered firstline therapy but mainly affect pain and stiffness, although
not significantly affecting the disease process. Therefore, disease-modifying antirheumatic drugs such as methotrexate are used early in the course of the disease, especially in polyarticular and systemic JRA. Intra-articular long-acting corticosteroid injections are used with increasing frequency to treat pauciarticular JRA and prevent flexion contractions and leg length discrepancies. Systemic corticosteroids are reserved for patients with severe systemic features of JRA or as a bridging medication until diseasemodifying drugs are effective. In recent years,
biologic-modifying medications acting on specific molecules in the inflammatory process have been developed for treating arthritis. Etanercept, a soluble receptor of tumor necrosis factor, has been approved for the treatment of polyarticular JRA not responsive to methotrexate. Adalimumab, a humanized tumor necrosis antibody has also been approved for use in polyarticular JRA. Infliximab, a mouse-based tumor necrosis antibody, may be effective for those patients with uveitis not responsive to methotrexate. Abatacept, a T-cell co-stimulator inhibitor was also approved for treatment of polyarticular arthritis. Anti-interleukin 1 and antiinterleukin 6 biologic medications may be effective, especially for patients with systemic JRA.
Physical and occupational therapy is also very important in maintaining joint range of motion and function. Physicians from other specialities participate in the treatment of JRA including ophthalmologists, orthodontists, and orthopedists. It is hopeful that with modern medical therapy less orthopedic intervention, particularly joint replacement surgery, will be necessary.
Prognosis
The prognosis varies with the subtype of JRA. Unfortunately, previously quoted statistics that as many as 80% of children will enter remission before adulthood are incorrect and are not supported by current data. Recent studies have shown that most children with polyarticular JRA and systemic or pauciarticular JRA with a polyarticular course will continue to have active disease as adults. Less than one third achieve long-term remission. Children having systemic-onset JRA with a persistent fever, high platelet count, and a polyarticular course and children with RF-positive JRA will have a particularly poor prognosis, with severe joint erosions and disability. These children require more aggressive therapy.