O2) to alveolar ventilation (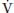 A):
A):
| (22.1) |
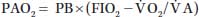
As far as  A is directly proportional to CO2 production (
A is directly proportional to CO2 production ( CO2),
CO2),
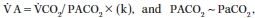
Equation 22.1 can be transformed into the alveolar gas Equation 22.2:
| (22.2) |
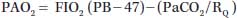
where RQ, respiratory quotient; RQ = 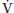 CO2/
CO2/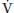 O2 (0.8 in resting state); and 47, saturated water vapor pressure at the PB in mm Hg.
O2 (0.8 in resting state); and 47, saturated water vapor pressure at the PB in mm Hg.
1. Respiratory depression (hypoventilation) can be either central (from CNS disease such as trauma, tumor, etc., or medications that cause respiratory depression such as narcotics or sedatives) or peripheral (from airway or chest trauma, neuromuscular disease, or muscle relaxants).
a. Central hypoventilation: Voluntary control of ventilation requires preserved consciousness and cerebral cortex and plays a minimal role in normal breathing. Involuntary motor control of ventilation is located in the respiratory center in the brain stem (pons and medulla). After receiving input from the central and peripheral chemoreceptors, baroreceptors (located in the vasculature), and stretch receptors (located in the bronchial tree and lung parenchyma), the respiratory center sends its output to the respiratory muscles via respiratory motor pathways. Input from the peripheral receptors is transferred to the respiratory center via the vagus nerve (Cranial Nerve X) from the aorta and glossopharyngeus (Cranial Nerve IX) from the carotid arteries.
Central chemoreceptors are located in the medulla and are sensitive to a change of pH in the cerebrospinal fluid (CSF) in response to changes in the PaCO2. There is an increase in ventilation when the PaCO2 is > 40 mm Hg and/or the arterial pH is < 7.4. Central chemoreceptors are responsible for about 85% of ventilatory effort. Peripheral chemoreceptors react to PaO2 with an increase in ventilation when PaO2 is < 60 mm Hg as well as to PaCO2 and pH.
Variations in both the depth and rate of spontaneous respiration occur in 60% of patients who have an isolated brain injury. Hypoventilation and hyperventilation are both pathologic respiratory patterns that can lead to hypo- or hypercarbia, hypoxemia, pH disturbances, and electrolyte imbalance. Five respiratory patterns have been described in patients who have TBI or brain tumors (Fig. 22.1). These respiratory patterns are rarely observed in practice, however, because of early endotracheal intubation and mechanical ventilation in the management of patients after CNS injury.
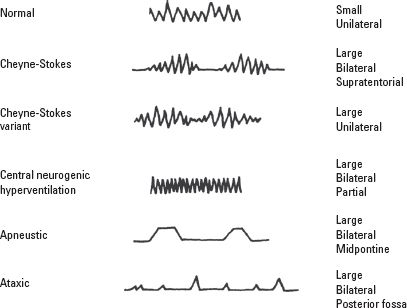
FIGURE 22.1 Respiratory patterns associated with closed head injury. Normal breathing in an awake patient who has a small, unilateral lesion. Cheyne-Stokes ventilation is a periodic breathing pattern in which hyperpnea alternates with apnea. The hyperpneic phase lasts longer than the apneic phase. The Cheyne-Stokes variant has a shorter apneic phase and is associated with large unilateral injuries. Central neurogenic hyperventilation is a sustained, regular, rapid, fairly deep hyperpnea. It occurs with a large pontine lesion, systemic hypoxia, or metabolic acidosis. It is often associated with brain stem tumors, including lymphomas and astrocytomas, and metastatic disease. Apneustic breathing occurs with large bilateral midpontine lesions. There is a brief 2- to 3-second pause at the end of inspiration, alternating irregularly with expiratory pauses. It has a very poor prognosis. Ataxic respiration is a pattern of irregular breathing in which shallow and deep breaths alternate randomly with irregular pauses. It is associated with medullary compression and has a very poor prognosis. The irregularity distinguishes it from Cheyne-Stokes ventilation. (Reprinted from Plum F, Posner JB. The Diagnosis of Stupor and Coma. 2nd ed. Philadelphia, PA: FA Davis Co., 1972 with permission.)
Commonly used anesthetic medications such as opioids, gamma aminobutyric acid (GABA)-ergic sedatives, and inhaled anesthetics inhibit central ventilation. The alpha-2 agonist dexmedetomidine lacks GABA-ergic or central effects. When used in sedative doses (0.2 to 0.6 mcg/kg/hour), dexmedetomidine causes a respiratory phenomenon that resembles natural sleep in which naturally occurring slight hypercarbia leads to hyperventilation with subsequent normalization of arterial PaCO2. As maintaining normocarbia is crucial in the management of patients who have brain and SCI, this unique feature of dexmedetomidine makes it a promising alternative to routinely used sedative medications in the management of neurosurgical patients in both the OR and the intensive care unit (ICU).
b. Peripheral hypoventilation
1. High cervical spinal cord lesions impair ventilation because of either partial or complete loss of innervation of the diaphragm (C3–C5), with loss of the ability to cough. The diaphragm is spared when the injury is below C6–C7. However, injury at the thoracic level results in the loss of intercostal and abdominal muscle function, which reduces FRC and creates paradoxical ventilation (chest retraction on inspiration and expansion during expiration), loss of cough, and reduced ability to clear secretions. All lung volumes are markedly decreased with the exception of residual volume.
With a C5–C6 lesion, ventilation may be inadequate and forced vital capacity (FVC) decreased to 30% of predicted capacity. Improvement of FVC occurs with time because of the strengthening of the accessory muscles (clavicular portion of the pectoralis major and sternocleidomastoid), development of spasticity (which stops the paradoxical chest wall motion), improvement of diaphragmatic function, and decreased use of narcotics and sedatives. At 5 months after injury, FVC reaches 60% of predicted values.
2. Associated injuries in multiple trauma patients including upper and lower airway injuries such as tracheobronchial fistula or disruption, rupture of the diaphragm, and thoracic trauma with rib fractures can all lead to acute hypoventilation and profound hypoxemia and hypercarbia.
2. Increased physiologic dead space is associated with drug administration (such as atropine and sodium nitroprusside for deliberate hypotension), mechanical ventilation with positive end-expiratory pressure (PEEP), hypovolemia, hypotension, hypocapnia, and pulmonary embolism. The Bohr Equation (22.3) can be used in ventilated patients to calculate the physiologic dead space:
| (22.3) |
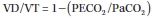
where VD, volume of the dead space; VT, tidal volume; PECO2, end-tidal CO2 tension in mm Hg, and PaCO2, arterial CO2 tension in mm Hg.
Please note that VD/VT represents a fraction of the dead space ventilation, which normally should be about 0.3 (or 30% of tidal volume) in mechanically ventilated patients.
3. Mechanical impairment. Airway obstruction from oropharyngeal soft tissue, uncleared secretions, hemoptysis, foreign bodies, bleeding, or bronchospasm can cause mechanical impairment of effective alveolar ventilation. The restriction of free chest movement from surgical positioning, surgical retraction of the chest and abdomen, traumatic rupture of the diaphragm, pneumothorax, hemothorax, flail chest, and restrictive lung disease can reduce alveolar ventilation significantly. Proper recognition and treatment of traumatic chest injuries are crucial to the successful management of patients after CNS trauma.
B. Causes of increased P(A-a)O2. The P(A-a)O2 may be increased as the result of both extrapulmonary and pulmonary factors. Decreases in cardiac output and either a respiratory or metabolic alkalosis are important extrapulmonary factors that increase P(A-a)O2 in the presence of lung disease. Dramatic increases in dead space ventilation, such as with massive pulmonary embolism, may lead to arterial hypoxemia as well. Pulmonary disorders can increase P(A-a)O2 because of shunting,  A/
A/  mismatch, and limitation of diffusion.
mismatch, and limitation of diffusion.
1. Extrapulmonary factors that commonly contribute to hypoxemia in neurosurgical patients who have pre-existing lung disease are hyperventilation to reduce ICP and excessive depletion of intravascular volume from fluid restriction and the administration of diuretics.
a. Hyperventilation to induce hypocapnia results in a respiratory alkalosis with a decrease in venous return and cardiac output. Hypocapnia increases pulmonary shunt, especially in the presence of lung disease. The mechanism is unclear but might be related to hypocapnic bronchoconstriction and inhibition of hypoxic pulmonary vasoconstriction. In addition, the oxyhemoglobin dissociation curve shifts to the left, resulting in a lower PaO2 for any given oxygen saturation (Fig. 22.2). This is especially noticeable when the PaO2 ≤ 70 mm Hg. Although helpful in reducing ICP because of cerebral vasoconstriction, hyperventilation should be limited to short periods with a PaCO2 of 30 to 35 mm Hg. A PaCO2 < 25 mm Hg is avoided because of the potential for worsening cerebral hypoxia and neurologic outcome through excessive vasoconstriction. Hyperventilation also decreases venous return, mixed venous oxygen tension (P O2), and cardiac output.
O2), and cardiac output.
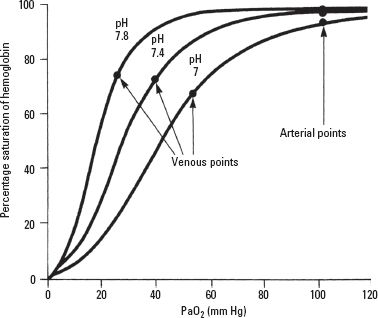
FIGURE 22.2 Shift in the oxyhemoglobin dissociation curve with change in blood pH.
b. Hypovolemia and decrease in P O2. PaO2 is lower when the P
O2. PaO2 is lower when the P O2 is decreased, as can occur with decreased cardiac output from hypovolemia, increased oxygen consumption, and severe anemia with increased oxygen extraction. The effect of P
O2 is decreased, as can occur with decreased cardiac output from hypovolemia, increased oxygen consumption, and severe anemia with increased oxygen extraction. The effect of P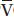 O2 can be quite significant, especially when there are areas of low
O2 can be quite significant, especially when there are areas of low 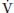 A/Q and shunt (Fig. 22.3). For instance, in the presence of
A/Q and shunt (Fig. 22.3). For instance, in the presence of 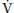 A/Q mismatch, PaO2 is 80 mm Hg when P
A/Q mismatch, PaO2 is 80 mm Hg when P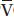 O2 is 50 mm Hg. If P
O2 is 50 mm Hg. If P O2 is decreased to 20 mm Hg from a decrease in cardiac output, then PaO2 decreases to 40 mm Hg without any change in the degree of
O2 is decreased to 20 mm Hg from a decrease in cardiac output, then PaO2 decreases to 40 mm Hg without any change in the degree of  A/
A/ inequality or shunt. Therefore, low cardiac output from hypovolemia or myocardial depression increases P(A-a)O2 and might result in hypoxemia, especially in the presence of pulmonary disease.
inequality or shunt. Therefore, low cardiac output from hypovolemia or myocardial depression increases P(A-a)O2 and might result in hypoxemia, especially in the presence of pulmonary disease.
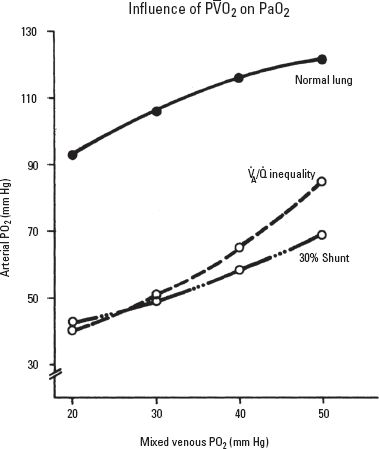
FIGURE 22.3 Influence of mixed venous oxygen tension on arterial oxygen tension. (Reprinted from Dantzker DR. Ventilation-perfusion inequality in lung disease. Chest. 1987;91:749–754 with permission.)
2. Pulmonary factors that increase P(A-a)O2 in critically ill neurosurgical patients can result from neurogenic causes such as neurogenic pulmonary edema (NPE), TBI- and SCI-induced 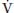 A/
A/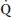 mismatch, and reduction in FRC. Abnormalities of lung parenchyma can also cause an increase in P(A-a)O2.
mismatch, and reduction in FRC. Abnormalities of lung parenchyma can also cause an increase in P(A-a)O2.
a. Neurogenic causes
1. Neurogenic pulmonary edema develops in severe CNS injury such as TBI, subarachnoid hemorrhage (SAH), intracranial hemorrhage (ICH), SCI, intracranial surgery, and conditions causing intracranial hypertension (tumor, hematoma, abscess). Of immediate onset, NPE becomes clinically recognizable 2 to 12 hours after injury. The pathogenesis of NPE has been suggested recently to involve “triggering” of the posterior hypothalamus and medulla to induce sympathetic stimulation resulting in the massive production of vasoconstrictors, such as norepinephrine, neuropeptide Y, and endothelin as well as proinflammatory mediators and nitric oxide (NO). Subsequently, an increase in pulmonary capillary pressure (transmural pressure > 40 mm Hg) causes endothelial and epithelial damage resulting in intra-alveolar leakage
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


