Properties, Absorption, and Disposition of Local Anesthetic Agents
Laurence E. Mather
Geoffrey T. Tucker
We dedicate this essay to the memory of our late friends and colleagues who contributed much to our knowledge of local anesthetic agents: Ray Fink (1914–2000), John Bonica (1917–1994), Bruce Scott (1925–1998), Ben Covino (1930–1991), Terry Murphy (1937–1996), and Anton Burm (1949–2005).
It is now more than three decades since the drafting of this chapter for the first edition of this book. In previous editions, the chemical evolution of local anesthetic agents from the ester caines of a century ago into the amide caines and chiral caines commonly used today was outlined as a significant part of anesthesia history. Throughout this evolution, the principle of promoting greater safety in regional anesthesia has not changed—and our dictum from previous editions thus remains unchanged:
The ideal use of regional anesthesia requires administration of sufficient local anesthetic agent to be effective but not so much so that toxicity develops. The anesthesiologist must have a thorough knowledge of the anatomic and physical landmarks to perform neural blockade in addition to a thorough knowledge of the pharmacology of the individual agents to be used. This includes familiarization with the disposition in the body of local anesthetic agents and, in particular, knowledge of their systemic absorption after the various methods of neural blockade.
The original evolution from prototypic cocaine to the myriad ester caines was driven by the clinical need to have an agent without the toxic and addictive potential of cocaine; the further evolution from ester caines to amide caines was driven by the pharmaceutical need for greater molecular stability. The need for safer, long-acting local anesthetic agents led to the more recent evolution to chiral caines, but it seems likely that continued evolution along the lines of existing chemical entities has now waned. It is, however, still important to examine relationships between the chemical and physicochemical properties of the agents and their fate in the body, and to delineate the role of pharmacokinetics in the overall response to regional anesthesia. Rather than provide a catalog of new and old data, we have continued to concentrate on the underlying principles of safe, effective anesthesia.
Structure of Local Anesthetics
Many chemically and pharmacogically diverse substances have local anesthetic activity, but remarkably few are used for clinical anesthesia. Conversely, the contemporary clinically useful local anesthetic agents (Table 3-1) have a variety of pharmacologic actions—some of which are exploited therapeutically, and others of which are avoided assiduously. These agents come from a small number of chemical families that still retain the clear ancestral line of cocaine, the original local anesthetic agent; however, paradoxically, cocaine can no longer be regarded as a typical local anesthetic agent, and it is rarely used as such.
Local anesthetic agents can be classified with respect to their chemical structures in two principal, interrelated, ways: by their functional groups and by their physicochemical properties. A comprehensive review of this interrelationship has been provided by Büchi and Perlia (1), albeit with many agents that are unknown except to those with a penchant for anesthetic history.
The functional group requirements for classical local anesthetic agents were established over 100 years ago with the discovery of the local anesthetic anesthesiophore (i.e., the smallest part of the cocaine molecule that retained local anesthetic activity) (2). It was soon realized that the anesthesiophoric part of cocaine was relatively simple and could be reduced to an aromatic (i.e., benzene ring–derived) head that conveys lipophilicity, an amino group that conveys hydrophilicity by way of its ability to form a charged species (or conjugate acid), and a linking chain that joins the two. The amino group was found to be optional, as demonstrated by the preparation of ethyl p-aminobenzoate (benzocaine) in 1890. The need for an injectable substance also required water solubility, and this was achieved and retained through an ionizable amino group, as in cocaine. Cocaine has an ester group in the linking chain, and this group was used in other agents until Löfgren’s research showed that it could be replaced with an isosteric (i.e., having similar spatial characteristics) amide group (3). By the late 1960s, local anesthetics were usually classified by their linking chain groups as ester or amide agents (4). As once-common ester-type local anesthetic agents are now used relatively rarely, this division has more historical significance than pharmacologic importance for contemporary anesthesiologists. A third classification, based upon stereochemistry, is now germane.
The predominant physicochemical properties are lipophilicity and the ionization constant (pKa)a of the amino group. Most other properties, such as affinity for macromolecules and water
solubility surface activity, as well as pharmacologic potency for both neural blockade and systemic toxicity, derive from these. Lipophilicity for both esters and amides alike is increased by alkyl and aryl substitution into the carbon framework of the molecule and/or on the amino functional group. The ionization constant determines the relationship between the fractions of local anesthetic agent molecules in the ionized (more water soluble, but more local anesthetically active) and nonionized (more lipophilic) states for any given environmental pH. Amine salts are used to produce water-soluble forms for injection: These are mostly hydrochloride salts (i.e., readily dissociating conjugate acids formed by the interaction of the amine with hydrogen chloride [HCl]), but sometimes other salts such as sulfates from sulfuric acid, acetates from acetic acid, and other compounds are used to achieve the required water solubility to produce the necessary local anesthetic concentration. On the premise that nonionized molecules are better able to cross (lipoidal) membranes, these properties determine much of the local and systemic pharmacokinetics and pharmacodynamics of the local anesthetic agents. Together, lipophilicity and hydrophilicity determine surface activity and the related tissue irritancy (5,6). Not surprisingly, tissue irritancy is used to screen out substances from further development as local anesthetic agents.
solubility surface activity, as well as pharmacologic potency for both neural blockade and systemic toxicity, derive from these. Lipophilicity for both esters and amides alike is increased by alkyl and aryl substitution into the carbon framework of the molecule and/or on the amino functional group. The ionization constant determines the relationship between the fractions of local anesthetic agent molecules in the ionized (more water soluble, but more local anesthetically active) and nonionized (more lipophilic) states for any given environmental pH. Amine salts are used to produce water-soluble forms for injection: These are mostly hydrochloride salts (i.e., readily dissociating conjugate acids formed by the interaction of the amine with hydrogen chloride [HCl]), but sometimes other salts such as sulfates from sulfuric acid, acetates from acetic acid, and other compounds are used to achieve the required water solubility to produce the necessary local anesthetic concentration. On the premise that nonionized molecules are better able to cross (lipoidal) membranes, these properties determine much of the local and systemic pharmacokinetics and pharmacodynamics of the local anesthetic agents. Together, lipophilicity and hydrophilicity determine surface activity and the related tissue irritancy (5,6). Not surprisingly, tissue irritancy is used to screen out substances from further development as local anesthetic agents.
Table 3-1 Physicochemical Properties of Local Anesthetics | |
|---|---|
|
Ester Caines
Cocaine was the first local anesthetic to be introduced into modern clinical medicine. Cocaine is an amine extracted from the leaves of the plant Erythroxylon coca and other plants of the same genus, native to Peru and Bolivia. During the 1850s, the plant was discovered in South America by German scientists and its principal active ingredient, the (tertiary) amine cocaine (“coca-ine”), was subsequently extracted, purified, and introduced into clinical medicine in the latter part of the 19th century. Final elucidation of the chemical structure of cocaine did not occur until 1924 but, by then, many hundreds of synthetic substitutes had already been prepared and trialed based upon the anesthesiophore of the cocaine molecule. The introduction of procaine (Novocaine)b in 1905 heralded a generation of a large family of ester caine local anesthetics having greater safety than cocaine and, most importantly, lacking its addictive/psychostimulant properties. Of these, chloroprocaine (Nesacaine) and tetracaine (amethocaine, Pontocaine) still remain popular with some anesthesiologists—the former for spinal anesthesia and the latter for topical anesthesia—along with some other ester caines, such as oxybuprocaine (Novesine) and proparacaine (Proxymetacaine), that are still used by some ophthalmologists.
As a class, ester caines in solution are subject to degradation by chemical hydrolysis, notably upon heat sterilization. This led to their replacement by amide caines that, as a class, resist chemical hydrolysis. The main pharmacologic benefit of ester caines, however, is that they are readily hydrolyzed metabolically by plasma and tissue esterases. Butamben, a benzocaine derivative of bygone years, received attention again in the late 1990s for its newly discovered ability to provide long-lasting and somewhat selective neural blockade after epidural administration of a suspension, but difficulties with its formulation appear to have precluded its continued development.
Amide Caines
The early 1930s saw the introduction of dibucaine (cinchocaine, Nupercaine), a quinoline derivative with the ester linkage replaced by a carbamoyl group. This agent is rather toxic, and its use was confined mainly to spinal anesthesia. It still provides the basis of a laboratory test of serum cholinesterase activity. The next major development occurred in two rival Swedish companies, AB Astra and AB Bofors, where some significant variations resulted in a new generation of amide caines. In these, the labile ester linkage was replaced by the chemically sturdier amide group, and this is further protected from chemical, but not enzymic, hydrolysis by steric hindrance from the 2,6-dimethyl groups on the aromatic (xylidine) ring. Lidocaine (lignocaine, Xylocaine, 1940s) was first, followed by mepivacaine (Carbocaine, 1950s), bupivacaine (Marcaine, 1960s), prilocaine (Citanest, 1960s), etidocaine (Duranest, 1970s), articaine (Carticaine, 1970s), ropivacaine (Naropin, 1980s) and levobupivacaine (Chirocaine, 1990s). In etidocaine, the carbon of the amide linking chain is ethyl substituted. In prilocaine, the 2,6-xylidine ring has been replaced by an o-toluidine moiety, and the amide linking chain is methyl substituted, thereby protecting the amide link from chemical hydrolysis; prilocaine also has a secondary rather than tertiary amino group, as in its near-relatives. Articaine, which was originally introduced for dental anesthesia and relatively recently into mainstream anesthesiology, differs from the other commonly used agents in that the aromatic head is a thiophene ring; it has an amide linking chain to the amino group, but it also has an ester group.
By the late 1970s, routine use of the ester caines had waned, and lidocaine and bupivacaine had become the de facto standards of shorter- and longer-acting agents, mepivacaine and prilocaine continued to have a smaller following among some anesthesiologists and dentists for selected procedures, and etidocaine had developed an unfortunate reputation for producing motor block that outlasted sensory block. At about the same time, concern over the safety of etidocaine and bupivacaine was becoming prominent (7,8).
As safety issues in regional anesthesia have continued to come to the fore, the future place of bupivacaine has now become questionable (9,10), especially with the introduction of ropivacaine and levobupivacaine, which have similar anesthetic activities but greater margins of safety (10,11). Articaine is now finding a wider place among anesthesiologists due to recognition of its relatively greater margin of safety (12).
Chiral Caines
The next major development came in the 1980s, with the development of ropivacaine—the first synthetic chiral caine—followed, a decade later, by the development of levobupivacaine.
Many substances, including a wide variety of anesthetic drugs (e.g., halothane, enflurane, isoflurane, desflurane, ketamine, thiopental, mepivacaine, bupivacaine, prilocaine, etidocaine, ropivacaine, and articaine) have a (single) chiral center (from the Greek, cheir, meaning “hand”). This center, also referred to as an asymmetric carbon atom or center of asymmetry, has four different functional groups bonded to it. This structure imparts a particular type of stereoisomerism known as enantiomerism. Where it is required to distinguish between them, substances lacking a chiral center (e.g., lidocaine, procaine, tetracaine and sevoflurane) may be referred to as achiral.
Enantiomers (also referred to as enantiomorphs) are stereoisomers having nonsuperimposable mirror image relationships (i.e., one enantiomer cannot be superimposed upon the other without chemical bonds being broken and remade differently).
Enantiomers (also referred to as enantiomorphs) are stereoisomers having nonsuperimposable mirror image relationships (i.e., one enantiomer cannot be superimposed upon the other without chemical bonds being broken and remade differently).
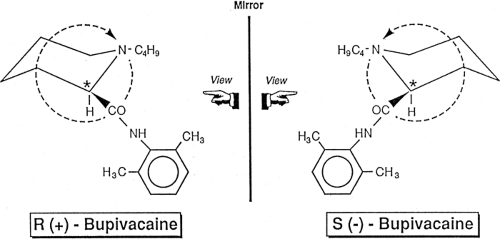 Figure 3-1. Application of the sequence rules of Cahn, Ingold, and Prelog to the enantiomers of bupivacaine. The chiral carbon is marked *. The smallest attached group, hydrogen, is projected away from the viewer; the other groups nitrogen (of n-N-butyl), carbon (of carbonyl), and carbon (of piperidine ring methylene) are arranged in clockwise order of decreasing size to give R-bupivacaine, which is dextro-rotatory; its enantiomer, S-bupivacaine, has the opposite order and is levo-rotatory. |
Enantiomers have identical physical and chemical properties, but differ in optical activity—the direction with which they rotate plane polarized light—a distinguishing property for which they became known as optical isomers, a now-obsolete term in the context of stereopharmacology. A single stereoisomer, free from its enantiomer, is referred to as enantiopure. A racemate (racemic mixture) consists of equal amounts of both enantiomers (and, therefore, has null rotation of plane polarized light); nonracemic mixtures consist of unequal mixtures of enantiomers. Racemization is the irreversible production of a racemic mixture from a nonracemic chiral starting material. Inversion is conversion of a chiral substance to its enantiomer. Enantioselectivity refers to a preference for one enantiomer over the other, whereas enantiospecificity refers to marked distinctiveness or exclusivity of that enantiomer. Diastereoisomers (or diastereomers) have multiple centers of chirality, and they can have multiple pairs of enantiomers (a pair for each chiral center), and normally differ in their chemical and pharmacologic properties (e.g., ephedrine and pseudoephedrine are diastereomers), and each consists of a pair of enantiomers.
Chiral drugs found in nature are usually single enantiomers because they are synthesized enzymically, and such reactions are usually stereospecific. Most synthetic chiral drugs are racemates, unless special synthesis is used to preserve the stereochemistry of a chiral center, or a chiral asymmetric synthesis is used to introduce a chiral center. Racemates are normally produced because there is an equal chance that both stereoisomers around a chiral center will be formed unless there is an energetic advantage for either one. Enantiomers behave essentially identically in an achiral environment, but in the chiral (amino acids, sugars, etc.) environment of the body, they behave as separate entities with potentially different pharmacokinetic and pharmacodynamic properties due to differences in their binding to macromolecules such as receptors (thus effect), membranes (thus distribution), and enzymes (thus metabolism) (13).
Three notations are used to describe chirality and associated optical activity, and all three appear in various publications about local anesthetic agents. First, (+) and (-), or dextro and levo (or d and l, now obsolete terminology) are associated with the pairs of enantiomers that rotate plane polarized light, respectively, to the right and left; that is, they are based upon optical properties (first noted in the 1830s). However, the direction of rotation is a phenomenologic feature only. A molecule may rotate plane polarized light in one direction when dissolved in one solvent but in the opposite direction in another solvent. Second, a systematic method of associating the stereochemistry with the direction of optical rotation was developed in 1919 by Emile Fischer and indirectly based upon structure. The Fischer-Rosanoff convention is based upon a molecule’s configuration relative to that of (+)-glyceraldehyde (now known to be (R)-2,3-dihydropropanal, see below), which was arbitrarily assigned a D configuration (its enantiomer, (-)-glyceraldehyde being designated L-glyceraldehyde), or relative to (-)-serine (L configuration). The configuration of a molecule would be assigned the D configuration if it (or a chemical degradation product that retained the chiral center) had the same direction of optical rotation as the model substance (+)-glyceraldehyde, and the L configuration if the direction of rotation was the reverse. The direction of rotation, as in D-(+)-bupivacaine, also may be added. The D and L notations are still used in sugar and amino acid chemistry. Third, in 1955, the sequence rules of Cahn, Ingold, and Prelog introduced a method for the unequivocal designation of molecular configuration by giving a sequence of priority to the four atoms or groups attached to a tetrahedral chiral center. With the smallest atom or group extending away from the viewer, the arrangement of the largest to smallest groups proceeding clockwise is designated as R– (for rectus); its enantiomer is designated S– (for sinister) (Fig. 3-1). If more than one chiral center is present, they must be designated by their configuration at each position. As examples, levobupivacaine, which has one chiral center, is (S)-1-butyl-2-piperidylformo-2′,6′-xylidide; natural (-)-cocaine, which has two chiral centers, the position of each being specified, is 3(S)-benzoyloxy-2(R)-methoxycarbonyl tropane.c
Contemporary literature still contains all three notations, depending upon the amount of information available and when the information was acquired, but only the Cahn-Ingold-Prelog notation denotes the absolute configuration. Racemates are designated (±)-, dl-, DL-, rac-, or RS-; nonracemic mixtures are designated by their enantiomeric excess (or ee), a measure for how much of one enantiomer is present compared to the other; for example, a sample with 40% ee in the R-enantiomer, would have the remaining 60% as racemic (with 30% of R-enantiomer and 30% of S-enantiomer), thus having a total of 70% R-enantiomer. The naming confusion is preserved by the use of international nonproprietary names such as levobupivacaine, which designates optical rotation rather than absolute configuration. The direction of optical rotation is only rarely of pharmacologic significance, so that it is often omitted from the name: (-)-(S)-bupivacaine is adequately designated S-bupivacaine (or by the nonproprietary name levobupivacaine). Application of the various forms of stereochemical notation is given in Table 3-2 using mepivacaine and bupivacaine as examples (522).
Table 3-2 Applications of stereochemical nomenclature to mepivacaine and bupivacaine | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Despite our modern attention to chirality, much was known about the chiral pharmacology of local anesthetics from work performed during the 1960s and 1970s, mainly at the Swedish companies AB Astra and AB Bofors. Prilocaine, mepivacaine, bupivacaine, etidocaine, and articaine were introduced into clinical use as racemates before it was appreciated how important these agents might turn out to be. (Bupivacaine, and the other racemic local anesthetics discussed here, were named originally to designate the racemate, so that there is no need to specify that the particular agent is a racemate unless required by the context. Once in the body, enantioselectivity in pharmacokinetics makes a racemic agent into a nonracemic mixture, so that a measured plasma concentration of bupivacaine, for example, will normally comprise an unequal mixture of R– and S-bupivacaine.) After the concern was raised about the disproportionate cardiotoxicity of bupivacaine, ropivacaine (which is alone among the synthetic local anesthetic agents to be produced as a single enantiomer from its inception) was announced (14), and levobupivacaine (enantiopure S-bupivacaine) was later introduced based upon findings of its lesser toxicity than either bupivacaine or its enantiomer dextrobupivacaine (R-bupivacaine) (15,16). Nonracemic mixtures also have been trialed, for example, with bupivacaine (17,18) but have not been developed commercially.
Although pharmacologic enantioselectivity was first recognized by Louis Pasteur in 1858, it is only relatively recently that the pharmacologic consequences of enantiomerism in the context of anesthesiology has been rediscovered (19,20,21,22,23,24). A variety of studies, mainly using bupivacaine enantiomers in different models and bioassays has, not surprisingly, shown that enantioselectivity at voltage-gated ion channel and other receptors mediating different neurogenic effects ranges from negligible to considerable (25,26,27,28,29,30,31,32,33,34,35,36,37,38). Presently, drug regulatory authorities are attuned to new chemical entity racemic drugs; these must be demonstrated as being no less safe than their enantiopure counterparts (39).
Analytical separation of local anesthetic (and many other) pairs of enantiomers for pharmacokinetic and other purposes is based on their different affinities for chiral macromolecular stationary phases, typically α1-acid glycoprotein (also known as orosomucoid), or cyclodextrin, packed into a chromatographic column (40,41,42). On a manufacturing scale, a racemate such as bupivacaine, an organic base, can be separated into its component R– and S-enantiomers by combination with a suitable enantiomer of an organic acid, such as tartaric acid, to form two diastereomer salts that have different solubilities and thus unique propensity for selective precipitation; this is a common laboratory process called resolution, and commercial methods have been patented for this process (43). Alternatively, chiral synthesis to preserve the stereochemistry at the chiral center of the reagents used can be used to produce the required enantiomer selectively (44). For any drug, the commercial method used ultimately depends on cost-effectiveness of the process and the chemistry of the particular drug.
Potency
An agent’s potency derives from its structural, stereochemical, and physicochemical properties: These properties govern the fraction of a dose reaching and residing at the relevant receptor, the agent’s affinity for the receptor, and its (biochemical and biophysical) efficacy once it has bound to the receptor.
The potency of local anesthetic agents is determined early in drug development by blockade of isolated nerve electrophysiologic studies and/or neural blockade procedures in intact laboratory rodents. Their relative potency is often specified in terms of their equi-effective anesthetic concentrations, and some well-known relationships between agents are given in Table 3-1. Equi-effectiveness is sometimes judged by clinical bioassay, for example, by subjects having similar characteristics of neural blockade (e.g., dermatomal–time diagrams) (45), and/or in amount of supplemental analgesic used by patients after surgery when the local anesthetic agent is used for some neural blockade procedure (46). Equal clinical effectiveness is, of course, a desirable standard of judgment, but it is a fairly imprecise standard in that, with reasonably similar agents being tested in typically small cohorts, most studies are more likely to fail to detect a difference between agents rather than show no difference between agents (type II error). More stringently, potency can be specified in terms of the (molar) dose and/or concentration of local anesthetic agent found in preclinical studies that is required to produce a defined extent and/or degree of neural blockade in a particular model or preparation, or the frequency of its occurrence in a certain population.
By-and-large, the same chemical and physicochemical changes that influence anesthetic potency among the various agents also influence potency for toxicity, whether the agents be ester- or amide-type, with some adjustments for differences introduced by also considering chirality. Among these agents, the more potent substances are more lipophilic and less hydrophilic, and this could engender situations in which the physical presentation of the local anesthetic agent is limited by the soluble amount of dose. Greater potency is also usually associated with a longer duration of neural blockade;
for example, intrathecal doses of bupivacaine 4- and 8-mg have approximately the same time courses of neural blockade as ropivacaine 8- and 12-mg (47).
for example, intrathecal doses of bupivacaine 4- and 8-mg have approximately the same time courses of neural blockade as ropivacaine 8- and 12-mg (47).
Various nerve blocks are now being used to provide a means for assessing the relative potency of local anesthetic agents for producing relevant effects (e.g., sensory or motor block) in a particular population (e.g., primiparous patients in first-stage labor). From studies using the “up-down” sequential single-dose technique, the median effective dose (ED50)d has been determined as a primary measure for comparing potency between agents or for assessing the usefulness of drug combinations (48,49,50). It has been found, for example, that the mean (and 95% confidence intervals) of the relative analgesic potency ratios in intrathecal labor analgesia were 0.65 (0.56–0.76) for ropivacaine-to-bupivacaine, 0.80 (0.70–0.92) for ropivacaine-to-levobupivacaine, and 0.81 (0.69–0.94) for levobupivacaine-to-bupivacaine, suggesting a potency hierarchy of spinal bupivacaine >levobupivacaine >ropivacaine (51). Similarly, in separate studies, the relative ropivacaine-to-bupivacaine analgesic potency ratio for epidural labor analgesia was found to be 0.6 (0.49–0.74), and that for ropivacaine-to-levobupivacaine potency, the ratio was 0.98 (0.80–1.20) (52,53). The different ratios for ropivacaine-to-levobupivacaine between intrathecal and epidural dosing presumably reflect the more complex milieu of epidural analgesia, as described later.
Some have criticized the use of ED50 as the appropriate measure of relative potency, because it represents only the 50th centile; that is, the mid-point of the dose-response curve, and that the ED95 would be a more useful measure for extrapolation to clinical practice (48,54,55). Some of the criticism is based on claims that the observed relative potencies of local anesthetics in clinical use do not fit with the observed ED50‘s. The ED95 is beyond the linearized (ED20 to ED80) portion of the slope of the (presumed sigmoidal) dose-response curve, so that the relationship between the ED50 and the ED95 need not bear the same dose proportionality relationship, which arises when comparing drugs with different slopes. Indeed, the slopes of the dose-response curves for different end points (e.g., sensory and motor blockade) may, and probably do, differ both between closely related agents, as well as within agents (56,57).
The up-down sequential dose method has also been used for assessing the effectiveness of treatments under particular conditions. For example, it has been shown that the ED50 for bupivacaine in late labor was greater by a factor of 2.9 (95% CI 2.7–3.2) compared with the MLAC in early labor (58). The up-down sequential dose method is also used assessing combination treatments with additives such as epinephrine or opioids (59,60). Others have also used the more formal isobolographic analysis with several combinations of doses for assessing the ED50 for combinations of drugs to determine whether a preferred dosage ratio exists (61).
Physicochemical Properties of Local Anesthetics
Basically, three mechanisms are involved in the movement of local anesthetic molecules within the body: bulk flow of the injected solution at the site of administration, diffusion into and through aqueous and lipoprotein barriers, and vascular transport from the site of administration. Of these, diffusion is most directly dependent on the physicochemical properties of the agent.
Table 3-3 Features common to most local anesthetics | |
|---|---|
|
According to Fick’s Law, the rate of passive diffusion (dQ/dt) of a drug through a biologic membrane at steady state may be approximated by Equation 1, in which D is the diffusion coefficient of the drug in the membrane; A and δ are, respectively, the area and the thickness of the membrane; K is the partition coefficient of drug between the aqueous and membrane phases; and ΔC is the concentration gradient.
Inasmuch as they determine D, K, and C, physicochemical properties will influence the rate of transport of local anesthetic agent at the membrane level, and potentially, therefore, the time course of anesthetic and pharmacologic effects. By influencing the equilibrium distribution of drugs between fluids and tissues, physicochemical properties also modulate activity and overall drug movement in the bloodstream.
Equation 1 can be simplified to Equation 2, in which the term comprising the constants D, A, K, and δ is combined into a permeability constant P; this equation indicates that the concentration difference across a membrane, ΔC, is the driving force for drug movement.
Some physicochemical properties of the clinically used local anesthetics are shown in Tables 3-1 and 3-3. As Table 3-1 shows, structural changes in the aromatic portion of the esters and in the amine group of the amides markedly alter physical properties such as oil or lipid/aqueous partition coefficients and affinity for proteins. These, in turn, have effects on potency, onset time, and duration of anesthesia. For example, with mepivacaine, substitution of a four-carbon n-butyl group for the one-carbon methyl group on the amine function gives bupivacaine a 35-fold increase in distribution coefficiente, increased protein binding, and increased potency. Lipophilicity is commonly estimated by the distribution coefficient with a suitable organic solvent and an aqueous buffer, typically at pH 7.4, with reference to the pKa (63). More polar solvents (e.g., chloroform)
produce larger values than do less polar solvents (e.g., heptane); n-octanol, an amphiphilic solvent that is now commonly used to represent biologic barriers, will dissolve nearly 5% water, thus offering a medium of appreciable solubility of the conjugate acid form of the local anesthetic bases (63,64,65). The pH – pKa relationship is relevant because it governs the ratio of concentrations of drug in the ionized (more water soluble) and nonionized (less water soluble) forms. The coefficients also may be calculated from chromatographic retention data (66). Differences in molecular weight among drugs from homologous series are usually also associated with parallel differences in lipophilicity and pKa making it difficult to isolate the effects of individual variables. Under these circumstances, differences in diffusibility and permeability occur as second-order effects.
produce larger values than do less polar solvents (e.g., heptane); n-octanol, an amphiphilic solvent that is now commonly used to represent biologic barriers, will dissolve nearly 5% water, thus offering a medium of appreciable solubility of the conjugate acid form of the local anesthetic bases (63,64,65). The pH – pKa relationship is relevant because it governs the ratio of concentrations of drug in the ionized (more water soluble) and nonionized (less water soluble) forms. The coefficients also may be calculated from chromatographic retention data (66). Differences in molecular weight among drugs from homologous series are usually also associated with parallel differences in lipophilicity and pKa making it difficult to isolate the effects of individual variables. Under these circumstances, differences in diffusibility and permeability occur as second-order effects.
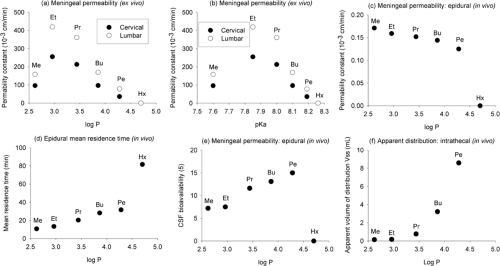 Figure 3-2. Composite figure showing the relationships between some physicochemical properties (lipophilicity given by octanol log P, and ionizability given by pKa) of n-N-alkyl homolog members of the mepivacaine family and some pharmacokinetic properties in models of spinal anesthesia. In an ex vivo model, A and B: Meningeal permeabilities. In an in vivo rabbit epidural model. C: Meningeal permeabilities. D: Epidural mean residence time as a measure of rate of drug dissipation. In an in vivo rabbit subarachnoid model, E: Bioavailability to CSF. F: Distribution within the subdural compartment. Me, methyl (mepivacaine); Et, ethyl; Pr, propyl (ropivacaine); Bu, butyl (bupivacaine); Pe, pentyl; Hx, hexyl. Compiled and redrawn from data in Clément R, Malinovsky JM, Hildgen P, et al. Spinal disposition and meningeal permeability of local anesthetics. Pharm Res 2004;21(4):706–716. |
Molecular Weight
Molecular weights of the commonly used local anesthetic agent bases vary from 220 to 288 Da (Table 3-1). These agents have essentially the same skeleton with the increases in molecular weight being due to alkyl substitution; hence, lipophilicity and pKa also change with molecular weight. Because the diffusion coefficient is inversely proportional to the square root of the molecular weight, and differences in molecular weight among related local anesthetic agents are relatively small, the diffusion coefficient does not contribute significantly to differences in their rates of diffusion. Changes in molecular weight produced by alkyl substitution, however, for example in pKa, are accompanied by changes in other physicochemical properties, and these potentially lead to other related effects (Fig. 3-2).
Ex vivo studies of the dural permeability of drugs have been performed with cadaveric tissues, and these demonstrate a relatively minor influence of molecular weight. Although a relatively simple model, cadaveric membrane-diffusion cell preparations are themselves subject to experimental vagaries of tissue choice (species, site and thickness, etc.) and preparation (notably fresh versus frozen) that probably explain some of the inconsistencies seen in data from different laboratories (67,68). Despite the simplicity, it is still difficult to identify the impact of the various factors and covariates (molecular weight and lipophilicity, for example, because these run together when a homologous series of drugs is used), and corrections need to be made for nonionized (c.f., total) concentration of drug (69). In vivo studies are even more difficult to interpret because of the competing influences of vascular activity, systemic absorption, and uptake into fixed spinal tissues, as well as cardiovascular sequelae of concurrent general anesthesia in the preparation of living subjects. These factors have not yet been investigated systematically, and it is only recently that microdialysis has provided a method for beginning to study in vivo dural permeability and to estimate the bioavailability to cerebrospinal fluid (CSF) of intrathecal and epidural local anesthetics (67,70,71,72).
When tested with an ex vivo preparation of monkey or rabbit spinal meninges, the permeability coefficients of a
heterogeneous series of drugs including lidocaine and bupivacaine were found to be independent of molecular weight (and molecular axis and molecular volume); however, a bell-shaped relationship was found with lipophilicity (as measured by the octanol-to-pH 7.4 buffer distribution coefficient) (73). When tested ex vivo with a series of homologous mepivacaine-like local anesthetics (i.e., progressively increasing molecular weight and lipophilicity), a parabolic relationship between permeability in lumbar and cervical meninges of the rabbit and lipophilicity was found. The permeability of human dura mater ex vivo to a variety of opioids and local anesthetic agents was found to depend on simple diffusion, independent of molecular weight, driven by the concentration gradient, and was not enantioselective (69,74,75). There is, however, some evidence to suggest that molecular weight might be relevant to the diffusion of local anesthetics in the sodium (Na+) channel of the nerve membrane (76).
heterogeneous series of drugs including lidocaine and bupivacaine were found to be independent of molecular weight (and molecular axis and molecular volume); however, a bell-shaped relationship was found with lipophilicity (as measured by the octanol-to-pH 7.4 buffer distribution coefficient) (73). When tested ex vivo with a series of homologous mepivacaine-like local anesthetics (i.e., progressively increasing molecular weight and lipophilicity), a parabolic relationship between permeability in lumbar and cervical meninges of the rabbit and lipophilicity was found. The permeability of human dura mater ex vivo to a variety of opioids and local anesthetic agents was found to depend on simple diffusion, independent of molecular weight, driven by the concentration gradient, and was not enantioselective (69,74,75). There is, however, some evidence to suggest that molecular weight might be relevant to the diffusion of local anesthetics in the sodium (Na+) channel of the nerve membrane (76).
Recent correspondence in the literature has also drawn attention to the relationship between molar and mass label quantities of some local anesthetic agents that could have implications for differences in dose requirements and the relative potency of similar agents (77,78,79). It is worth reiterating here some elements of that discussion because it also involves international differences that can occur in labeling requirements.
Local anesthetic agents are normally prepared as %weight/volume solutions of the hydrochloride “salts” of the relevant base, such that the molecular weight is equal to the base + HCl (= 36.5 Da) if anhydrous, additionally + H2O (= 18 Da) if monohydrate, etc. An aqueous solution for injection will be made of the salt, but the concentration of active ingredient may be specified either as that of the salt or the base; the water of crystallization is normally allowed for and adjustments are made. The relative concentrations of bupivacaine and levobupivacaine have the same molecular weight (= 288 Da), but whereas the concentration of bupivacaine is specified as the HCl salt (= 324.5 Da anhydrous), that of levobupivacaine is specified as the base (= 288 Da), at least for the European regulatory authorities. Thereby, the mass concentration of levobupivacaine HCl to make the solution has to be greater by 12.6% to achieve the same molar concentration of base as in a solution of bupivacaine HCl. Levobupivacaine could, therefore, appear to be more potent than bupivacaine if this difference in concentration were to be ignored. The message is plain: check the locally issued product label/information as to how the concentration of local anesthetic agent is specified!
Ionization
The inclusion of an amino group in the structure of most local anesthetics confers upon them the “split personality” of a weak base, meaning that they exist in solution partly as the nonionized free base and partly as the ionized cation (conjugate acid) (Equation 3):
Accepts proton (association):
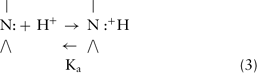
The position of equilibrium depends on the dissociation constant (Ka) of the conjugate acid and on the local hydrogen ion concentration (Equation 4). Thus,
where the square brackets indicate concentration or, more properly, activity. By rearranging Equation 4 and taking logarithms, the Henderson-Hasselbalch equation (Equation 5) is obtained:
where pKa is defined, by analogy to pH, as the negative logarithm of the dissociation constant of the conjugate acid under particular conditions of solvent and temperature (82,83,84).
Equation 5 shows that the pKa is equal to the pH at which the local anesthetic is 50% ionized, that is, when
The greater the pKa of a base, the stronger is that base—that is, it has a greater ability to attract a proton, and the smaller is the proportion of nonionized form at any pH. The ester-type agents have higher pKa values (8.5–9.1) than the amide types (7.6–8.2) (Table 3-1), which accounts, in part, for relatively poor penetrance and the need to inject these agents close to neural tissue. The effect on pKa values of differences in structure within the two main types of agents is complex, involving steric factors as well as the inductive effects of alkyl substituents on the amine nitrogen. For example, the greater pKa of bupivacaine compared with mepivacaine is explained by the effect of greater alkyl substitution, making the nitrogen atom relatively more negative (and able to attract a proton). In contrast, the lower pKa of etidocaine and of prilocaine reflects the effect of alkyl substituents on the bridging carbon atom in sterically hindering the approach of hydrogen ions to form the conjugate acid and in decreasing stabilization of the resultant cation by hydration.
Ionization is relevant to the solubility and activity of local anesthetics (Table 3-1) and their equilibrium distribution in various body compartments. Because the ionized forms are much more water-soluble than the free bases, the drugs are dispensed as their HCl salts, producing acidic solutions. This also helps to stabilize the esters since they are more readily hydrolyzed in alkaline conditions. The drugs are much more lipid-soluble in their free-base forms than in their conjugate acid forms. Thus, the nonionized fraction becomes essential for passage through lipoprotein diffusion barriers to the site of action on the nerve membrane. The balance between ionized and nonionized forms is shifted in favor of nonionized by the addition of bicarbonate (or other base), at the expense of decreased water solubility and the risk of reducing the effective drug concentration by drug precipitation (85).
Grouls et al. determined the effect of pH on the net flux across human meninges in vitro of lidocaine and bupivacaine. They found that the values at pH 4.0 were less than 20% that of the values measured at pH 7.4 (86). Decreasing ionization by alkalinization of the solution for injection will effectively raise the initial concentration gradient and flux (mass transferred per unit time) of diffusible drug (87,88) thereby increasing the rate of drug transfer. This maneuver has been successful in decreasing the latent period of a peripheral nerve block (89,90,91) but has not been universally successful for central neural blockade (92,93,94,95). Once at the nerve membrane, ionization is
again necessary for complete anesthetic activity (96). Increasing the pH of aqueous local anesthetic solution also increases the surface tension (97), and this has been shown to decrease the drop rate when lidocaine solution was being delivered under gravity feed.
again necessary for complete anesthetic activity (96). Increasing the pH of aqueous local anesthetic solution also increases the surface tension (97), and this has been shown to decrease the drop rate when lidocaine solution was being delivered under gravity feed.
The aqueous phases on either side of many body membranes differ in their pH values. Consequently, although nonionized drug concentrations in these phases will be the same at equilibrium, different concentrations of ionized and, therefore, of total drug will exist on either side of the membrane if a pH gradient exists. For a weak base, the equilibrium ratio (R) of total drug concentration across a membrane is given by Equation 6. Thus,
where the subscripts 1 and 2 refer to the two aqueous compartments. Total drug concentration will be greater in the compartment having the lower pH because a greater proportion will be in the ionized form. Thus, for example, lowered pH owing to infection in tissues surrounding a nerve results in less nonionized drug, which is the form that can cross the axonal membrane. Similarly, local anesthetics will diffuse from blood to acidic gastric contents, thereby maintaining a concentration gradient due to “ion trapping.”
Aqueous Solubility
The aqueous solubility of a local anesthetic is related directly to its extent of ionization and inversely to its lipid solubility (Table 3-1; Fig. 3-3). Despite quite large differences in the lipid solubility of the amide local anesthetic bases, the differences in aqueous solubility of the conjugate acids are smaller. Benzocaine and butamben, which lack an (ionizable) amino group, are almost insoluble in water. For this reason, their use is essentially confined to topical anesthesia, or injection for prolonged neural blockade after solubilization with agents such as dextran or suspension in polyethylene glycols. Mixtures of local anesthetic agents with excess bicarbonate will lead to precipitation of the local anesthetic base after the solubilizing acid (normally hydrochloric acid) has been neutralized by the inorganic base.
A low aqueous solubility was thought to be a limiting factor when selecting an agent for subarachnoid block, principally due to the risk of the agent precipitating in CSF and causing neural irritation. It is known that the aqueous solubility is less with more lipophilic agents, and solubility decreases with increasing pH. Transient neurologic disturbances after spinal local anesthetic administration has been reported sporadically for many decades. However, the problem has not vanished by substituting more dilute lidocaine or mepivacaine solutions, suggesting that water solubility of the local anesthetic is not the most critical factor (98,99). Determination of the roles of a particular local anesthetic agent, the particular method of pharmaceutical preparation, and/or any additives have been the subject of much research, but relatively little sound science has emerged (100). How much is due to the local anesthetic, its concentration, and its properties such as surface activity, or the procedure itself, remains controversial (101,102).
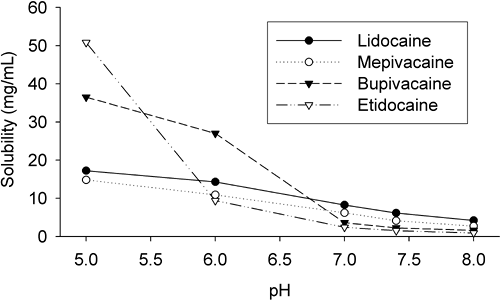 Figure 3-3. Aqueous solubility of some selected local anesthetics as a function of pH (Sorensen’s phosphate buffer). (Unpublished data from Mather LE.) |
Various experiments have been performed with local anesthetics to improve their water solubility, some with pharmaceutical manipulation to affect their local disposition, and some with alterations to the chemical structure. Whether these become useful clinically remains for future studies.
Lipid Solubility
The distribution coefficients of drugs measured in organic solvent/aqueous buffer systems in vitro are commonly used to reflect their relative in vivo distribution characteristics or lipophilicity (83,525). The values obtained normally differ quantitatively between systems due to both the polarity of the solvent and the extent of drug ionization at the chosen pH, such that their best use is in making comparisons between drugs within the same system (522). Net lipid solubility is independent of ester or amide grouping. Tetracaine (amino ester) is regarded as being highly lipid-soluble, as are ropivacaine, bupivacaine, levobupivacaine, and etidocaine (amino amides). Rosenberg et al. (103) have shown good rank order correlation between the n-heptane/buffer distribution coefficients of bupivacaine, etidocaine, ropivacaine, and lidocaine and their in vitro distribution into rat sciatic nerve and human epidural and subcutaneous fat. The intrinsic potencies (reciprocals of ED50) of amino ester, amino amide, and piperidine amide local anesthetics have been found to be proportional to their calculated solubility in nerve fibers as determined from their lipid solubility (104). However, the order breaks down when a substance has too great a lipid solubility, and it is suggested that a limiting octanol distribution coefficient of about 5 pertains to useful membrane penetration. This is shown in Figure 3-2, where the n-hexyl derivative of mepivacaine fails to significantly penetrate the meninges after epidural administration in rabbits. After lipid solubility reaches this critical level, the apparent anesthetic potency, along with the intrinsic systemic toxicity, decreases (producing bell-shaped curves). This is caused by the drug so favorably distributing into lipoidal barrier membranes that insufficient partitions move into axoplasm (to produce neural blockade) as well as vital organs (producing lethal effects). Nevertheless, local tissue toxicity (cell leakiness) increases continuously with increasing lipid solubility throughout the test series of drugs (79,105).
The relationships for the three classes of agent are essentially parallel but, for the same degree of calculated solubility of the local anesthetic base, the intrinsic potencies were found to be in the order amino ester >amino amide >piperidine amide (106). However, the relationship between potency and physicochemical properties is complex. The distribution coefficients are the result of the composite chemical group effects. Different relationships might apply in vivo, especially for
chiral agents. The relative proportions of base and conjugate acid of the individual agents depends on their pKa. Most relationships are developed in terms of base concentrations only when it is known that both forms are required for membrane stabilizing effect. Overall, a high lipid solubility would be expected to promote drug entry into membranes by increasing the diffusion rate, but this has to be balanced with a high fraction of drug in the nonionized state (106,107,108). The net effect on onset of maximum anesthetic action is difficult to predict, however, because a faster rate of diffusion is offset by a greater capacity for uptake into a membrane.
chiral agents. The relative proportions of base and conjugate acid of the individual agents depends on their pKa. Most relationships are developed in terms of base concentrations only when it is known that both forms are required for membrane stabilizing effect. Overall, a high lipid solubility would be expected to promote drug entry into membranes by increasing the diffusion rate, but this has to be balanced with a high fraction of drug in the nonionized state (106,107,108). The net effect on onset of maximum anesthetic action is difficult to predict, however, because a faster rate of diffusion is offset by a greater capacity for uptake into a membrane.
It has been found that high lipophilicity alone also does not predict the relative rate of transfer of local anesthetics and chemically similar drugs through meninges in ex vivo models (73). The ex vivo apparent permeability coefficients of homologous N-alkyl piperidine amides and a series of heterologous amide and ester caines follow a bell-shaped curve with increasing lipophilicity; their in vivo rate of absorption from site of administration in the epidural space to CSF constantly decreases with increasing lipophilicity, but their fractional availability to CSF increases (64,67). This apparent paradox reflects the relative balance in competing routes of local distribution into local fixed structures and clearance by local vasculature. Further complexities pertain even within homologous series. Because pKa values also increase with increasing alkyl substitution, the nonionized fraction increases significantly. Moreover, even when lipid solubility and nonionized fraction are taken into account, the intrinsic potency of the agents is subject to differences in preferred molecular geometry for fit to the local anesthetic receptor site (108). However, it is generally found that by promoting interaction with the hydrophobic components of receptors, a high lipid solubility will increase potency and duration of effect.
Protein Binding
Most drugs reversibly bind to/associate with proteins (and various other macromolecules) to some extent. This association, when it occurs within the body, can provide a transport medium and/or a temporary repository for the drug. Drug binding to proteins is a secondary property originating from physicochemical and stereochemical properties that has received much attention with local anesthetic agents.
Thermodynamic characterization of drug binding treats it as an adsorption process and involves the molar concentration of binding protein(s), the molar concentration of binding sites (or number per molecule of protein), and the association/dissociation constants of the interaction. Pharmacologic characterization, however, is more concerned with determining which protein(s) are involved, and the extent of binding at various drug and protein concentrations. If more than one drug and/or protein is involved, as typically occurs with drug binding in plasma, the relative extent(s) of binding will be governed by the (competitive) relative affinity constants of the separate drug–protein interactions.
Besides being more lipophilic, the longer-acting local anesthetics also exhibit higher degrees of binding to plasma and tissue proteins (Table 3-1), with greater plasma binding of the agent in its base form at higher pH values (109,110,111,112,113,114). This suggests that the binding forces are predominantly hydrophobic. Steric factors also apply, so that the enantiomers of chiral local anesthetics usually exhibit differences in degree of binding to proteins, although the differences are usually smaller than those between agents where differences in lipophilicity predominate. Differences between the enantiomers of some drugs may be large enough to account for differences in some receptor-related effects (e.g., for toxicity) or in pharmacokinetics (e.g., for distribution and/or metabolism).
Adsorption of local anesthetics to binding sites, or solubility within membranes or tissues, although producing relatively high apparent partition coefficients, may result in slower net penetration rates. This may be considered either as a lowering of diffusion coefficient or as a decrease in ΔC, the effective concentration gradient of diffusible drug (Equation 2). Binding of the drugs to proteins associated with the aqueous phases on either side of a membrane will affect the transfer and equilibrium distribution of total drug, analogously to ionization. Only the unbound drug will diffuse readily and, again, this will modify net drug transfer rate by an influence on ΔC.
Drug Absorption and Disposition
On the basis of an appreciation of the different structures and physicochemical properties of local anesthetics and the expected consequences of these differences, it is possible to inquire more specifically into their absorption and disposition in various parts of the body. In doing so, the fate of the agents is conveniently divided into consideration of their local disposition, systemic absorption, and systemic disposition and regional disposition (Fig. 3-4). The term disposition has a special meaning; it refers collectively to the processes of drug distribution into and out of tissues, and drug elimination by excretion and/or metabolism, while specifically excluding the process of absorption into the bloodstream.
Local Disposition
In contrast to many drugs, the primary effects of local anesthetics at both the pharmacologic level (neural blockade) and clinical level (analgesia and anesthesia) can be measured fairly objectively. The anesthesiologist is concerned particularly with the onset, spread, quality, and duration of block of one or many nerves with differing morphology. Ultimately, however, these variables depend on the drug’s concurrent distribution at, and dissipation from, the site of injection. Therefore, it would be valuable to have chemical measurements of the agents at these sites as a function of time in order to establish pharmacodynamic relationships. Regrettably, such data remain sparse, even from animals. Therefore, knowledge of the local disposition, or neurokinetics, of local anesthetics remains largely theoretical and is deduced from observation of the spatiotemporal changes in anesthetic effect, rather than from direct measurements of intraneural and perineural drug concentrations. Most data have been obtained from studying a particular agent under particular conditions. Despite these limitations, it is possible to draw some general conclusions.
Competing factors that affect local disposition of local anesthetics include dispersion by bulk flow of the injected solution, diffusion, and binding and vascular uptake of the agent (or relevant solute). Local blood supply is critical, as this is responsible for local clearance/systemic absorption of drug; metabolic breakdown seems less important, and is probably negligible with amide-type local anesthetic agents. It has been suggested that the local blood supply is responsible for delivery of drug to deeper structures of the spinal cord via the radicular arteries (115,116) but this is now considered unlikely (117). In addition to these factors, the pharmaceutic presentation of the agent can
be manipulated so that the rate of release of agent controls the balance between local and systemic uptake of the agent.
be manipulated so that the rate of release of agent controls the balance between local and systemic uptake of the agent.
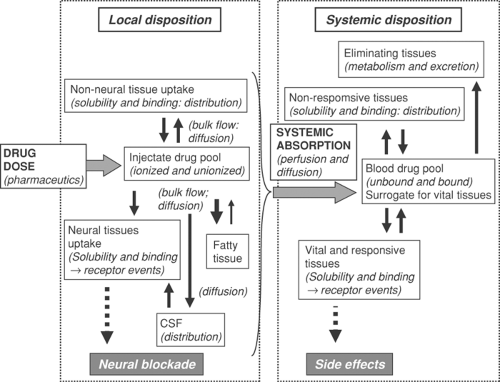 Figure 3-4. A conceptual model depicting the fate of local anesthetics injected perineurally. |
Although lipophilicity is an intrinsic physicochemical property of a drug resulting from its molecular structure, the effective lipophilicity can be altered by pharmaceutic presentation. A variety of drugs have been formulated with innovative methods to increase their local concentrations and/or residence times. Some examples include cyclodextrins to increase the local concentration of local anesthetics by making them more hydrophilic, and liposomes to make them more lipophilic. Both formulations have been found to be longer-acting after subarachnoid administration in laboratory animals than the simple aqueous solutions of the same drugs. The mechanisms of prolongation of action are presumed to differ but have in common the alteration of the rate of drug diffusion. Drugs combined with cyclodextrins are believed to have a slower rate of vascular absorption than aqueous solutions. Liposome or polylactide microsphere encapsulation provides a depot from which drug is released slowly. Liposomal preparations of local anesthetic agents have both a longer duration of action and a lower propensity for systemic toxicity than equivalent doses in aqueous media injected perivascularly, epidurally, or intrathecally in experimental animals (118,119,120,121) and in epidural, intercostal, or subcutaneous anesthesia in humans (122,123,124). Moreover, liposomal preparations of local anesthetic agents have a higher rate of penetration of the dermis than equivalent aqueous solutions (125).
Bulk Flow
The extent of spread of the injected solution might be expected to depend on its volume, the force (rate) with which it is injected, the turbulence generated, the size of the injected space, the physical resistance offered by tissues and fluids, gravity, the position of the patient during and after injection, and the resultant pressure gradients exerted by these factors. Bulk flow of local anesthetic solutions has been assessed by the use of marker dyes, radiographic contrast media, external counting techniques, and spinal canal models (126,127,128,129). Although the spread of analgesia may also indicate the extent of bulk flow, these are not necessarily equivalent because spread of effect is also determined by solute diffusion and local perfusion. For example, the observation that the epidural injection of bupivacaine in glycerol produced longer duration of analgesia than when injected in normal saline was interpreted as being due to a bupivacaine depot (130). Although this may be so, it also may be that the higher viscosity of the glycerol solution reduced the rate of bulk flow, thereby increasing the concentration of bupivacaine diffusing into the nerve structures. Indeed, the presence of fibrous septa within the injected spaces may create compartmentalization of perivascular spaces, thereby becoming the ultimate determinants of the observed effect by affecting the balance between bulk flow of local anesthetic solution and diffusion of local anesthetic agent (131).
Diffusion
Once the local anesthetic has been deposited and has spread physically in the extraneural fluids, it finds its way to sites of action in and on the nerve membrane largely by the process of diffusion (see Chapter 2).
The pattern of drug distribution within the cord is a complex function of accessibility by diffusion from the CSF, the relative myelin (lipoidal) content of various tracts, and the rate of drug removal by local perfusion. Recent studies in pigs, using opioids and microdialysis to sample the epidural space and CSF to perform a kinetic analysis, indicated complexity of the spinal pharmacokinetics of these drugs. The results with a series of opioids that have similar physicochemical properties to local anesthetic revealed that the bioavailability to the CSF and epidural space is determined primarily by their hydrophobicity, with less-hydrophobic drugs having greater bioavailability
(132). Other studies in animals using radiolabeled local anesthetics have shown their accumulation along the posterior and lateral aspects of the spinal cord as well as in the spinal nerve roots, but not to such a degree in the dorsal root ganglion or in the more central parts of the cord. Uptake of drug is higher in the gray matter than in the white matter of the cord, and posterior nerve roots had higher concentrations than anterior roots. Overall, the working model (Fig. 3-4) may be viewed as a reasonable representation of the central neuraxial fate of local anesthetics. By altering the ratio of base and conjugate acid, diffusion of local anesthetic agents can be influenced by a pH gradient, and differences in diffusion of local anesthetics can cause differences in neural penetration, thereby affecting differential block.
(132). Other studies in animals using radiolabeled local anesthetics have shown their accumulation along the posterior and lateral aspects of the spinal cord as well as in the spinal nerve roots, but not to such a degree in the dorsal root ganglion or in the more central parts of the cord. Uptake of drug is higher in the gray matter than in the white matter of the cord, and posterior nerve roots had higher concentrations than anterior roots. Overall, the working model (Fig. 3-4) may be viewed as a reasonable representation of the central neuraxial fate of local anesthetics. By altering the ratio of base and conjugate acid, diffusion of local anesthetic agents can be influenced by a pH gradient, and differences in diffusion of local anesthetics can cause differences in neural penetration, thereby affecting differential block.
Although stereochemical interactions between drugs and biomembranes may occur, the rates of diffusion of bupivacaine enantiomers across isolated meningeal membrane were found not to be enantioselective (69,75). Others have produced evidence for a pharmacokinetic explanation for tachyphylaxis after peripheral intra- or extraneural sciatic or subcutaneous administration of (pH 7.0 adjusted) lidocaine in rats. They found that progressive tachyphylaxis to nociceptive stimuli was associated with diminished respective neural or skin concentrations of lidocaine and claimed a pharmacokinetic (diffusion) basis for tachyphylaxis (133), but local tissue pH, unfortunately, was not measured as a variable.
pH Effects
Any factor that creates local extracellular acidosis should retard net diffusion of local anesthetic to the nerve by increasing and sustaining drug ionization (see Equation 5), and this was demonstrated by Ibusuki, et al. (134), using procaine and crayfish giant axons. A low pH may be preexisting, for example, as a result of infection, or may be induced by injection of the anesthetic solution. A number of studies have indicated that raising the pH of local anesthetic solutions to near neutral often hastens the onset of effects, sometimes prolongs the duration of effects, and usually decreases the local irritation or pain on injection. Capongna et al. (93) found that alkalinization produced the best results with lidocaine and bupivacaine for epidural block, with lidocaine for brachial plexus block, and with mepivacaine for sciatic and femoral nerve blocks. Such effects are not inevitable, however, and shortened onset was not demonstrated with local anesthetic mixtures used for ophthalmic sub-Tenon block or lidocaine for brachial plexus anesthesia (135,136,137).
Differential Block
The propensity to produce a marked differential blockade is a characteristic of some long-acting local anesthetic agents, particularly tetracaine and etidocaine, which produce profound motor blockade; bupivacaine provides a balance of analgesia and motor loss, whereas ropivacaine and levobupivacaine can provide analgesia with a minimum of motor loss (46,138,139). In the case of epidural block, Bromage (140) suggested that this phenomenon may be due to deeper penetration of the spinal cord by more lipid-soluble agents causing interference with long descending motor pathways. Bupivacaine, however, is more lipid-soluble than tetracaine and lidocaine, which produce less differential sensory loss, and the differences in sensory–motor dissociation seen with the various agents are also apparent after peripheral nerve block procedures, in which penetration of the cord is not a factor (141). To accommodate the latter, differences in the ability of the agents to diffuse into individual sensory and motor fibers have been postulated (106). Thus, from electrophysiologic studies with isolated rabbit vagus nerve, Gissen et al. (142) concluded that, compared to etidocaine, less lipid-soluble bupivacaine diffuses relatively slowly into fast-conducting A (motor) fibers at low concentrations. This is supported by other electrophysiologic studies (143), although Fink and Cairns (144) discount diffusion within a nerve as a contributory factor to differential block. Nevertheless, when comparisons were made between a wide variety of agents, it was found that in vitro blockade of A-fibers occurred before that of C-fibers with procaine, ropivacaine, and bupivacaine and vice versa with tetracaine, etidocaine, and mepivacaine; blockade occurred at the same time with lidocaine. In summarizing the results of their studies, Wildsmith et al. (106) noted that equipotent concentrations of the various drugs blocked C-fibers at the same rate but that blockade of A-fibers was more dependent, as expected, on the physicochemical properties (see Chapter 2).
Metabolism
Local metabolism, if it occurs, has a negligible influence on the neurokinetics of local anesthetics and the time course of conduction blockade.
Systemic Absorption
Local anesthetics produce their intended effects at or near their site of deposition, without requiring transit through the blood to a blood-perfused “effect site.” Indeed, their presence in the circulation is normally undesirable, being associated with lessened intended actions and an increased risk of systemic toxicity. Used in combination with information about putatively “toxic” blood (or plasma or serum) concentrations of local anesthetic agents, knowledge of their systemic absorption helps to set confidence limits on the likelihood of a systemic toxic reaction after various block procedures. This, apart from curiosity or regulatory requirement, is the main reason for measuring their blood concentrations. Indirectly, appreciation of rates of systemic absorption can suggest also the relationship between block and the amount of drug remaining at the site of injection, and this information can be useful in assessing other relevant phenomena, such as the relative potency of the drugs.
In humans, measurement of drug concentration–time profiles in the peripheral circulation has been widely used to assess systemic uptake of the different agents after virtually every neural blockade procedure. Tables of these data have been published in many reviews (145,178). Because these profiles are the net result of both systemic absorption and disposition, they are of value mainly to determine relative changes or differences in drug uptake after different agents, conditions or procedures. The maximum plasma drug concentration (Cmax) and the time of its occurrence (Tmax) are, therefore, not absolute indicators of the absorption kinetics of local anesthetics administered perineurally. Nevertheless, Cmax and Tmax, when measured under comparable conditions, are useful in making clinically relevant (but superficial) comparisons between drugs, procedures, and/or subjects, but they must be qualified as to whether they were derived from arterial or venous blood samples, and if venous, which venous. Arterial blood drug concentrations are essentially the same at all sampling points. Venous blood drug concentrations normally differ according to local tissue morphology, venous tone, perfusion, and neural blockade, because the net rate of drug equilibration within the tissues is affected by these factors. Central venous blood drug concentrations resemble arterial concentrations, but are subject to modulation by drug uptake in the lungs.
In general, Cmax in arterial blood for a particular local anesthetic and block procedure will primarily depend on the total
dose (Figs. 3-5 and 3-6). As a rule, a larger Cmax and a shorter Tmax will occur from drug administration into regions of higher perfusion. In general, the time of greatest risk of systemic toxicity coincides with Tmax in arterial blood, and this will vary from about 5 to 45 minutes; Tmax for peripheral venous blood will lag somewhat and vary from about 15 to 90 minutes (Fig. 3-7), with the values being mainly related to the speed of injection (147), drug and site of injection (148,149), and often with inexplicable variability between subjects for a given treatment (150). Note that Tmax can be a fairly imprecise variable for comparing between studies as it also depends to some extent on the frequency of blood sampling.
dose (Figs. 3-5 and 3-6). As a rule, a larger Cmax and a shorter Tmax will occur from drug administration into regions of higher perfusion. In general, the time of greatest risk of systemic toxicity coincides with Tmax in arterial blood, and this will vary from about 5 to 45 minutes; Tmax for peripheral venous blood will lag somewhat and vary from about 15 to 90 minutes (Fig. 3-7), with the values being mainly related to the speed of injection (147), drug and site of injection (148,149), and often with inexplicable variability between subjects for a given treatment (150). Note that Tmax can be a fairly imprecise variable for comparing between studies as it also depends to some extent on the frequency of blood sampling.
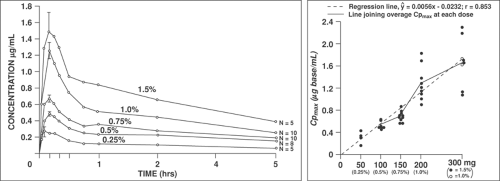 Figure 3-5. A: Plasma etidocaine concentration–time relationships after intercostal block in surgical patients with different concentrations (achiral assay). B: Maximum plasma etidocaine concentrations from the study in A showing the relationship of Cmax to dose. Compiled from Van de Velde M, Dreelinck R, Dubois J, et al. Determination of the full dose–response relation of intrathecal bupivacaine, levobupivacaine, and ropivacaine, combined with sufentanil, for labor analgesia. Data from Bridenbaugh PO, Tucker GT, Moore DC, et al. Preliminary clinical evaluation of etidocaine (Duranest): A new long-acting local anesthetic agent. Acta Anaesthesiol Scand 1974;18:165. |
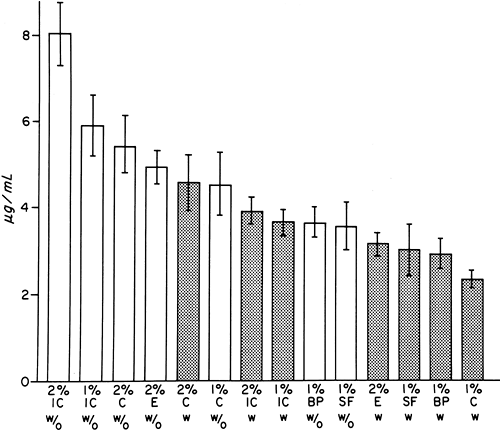 Figure 3-6. Systemic absorption of mepivacaine (achiral assay) in surgical patients after various regional block procedures, as indicated by the mean Cmax ± SEM of the values. IC, intercostal block; C, caudal block; E, epidural block; BP, brachial plexus block; SF, sciatic/femoral block; w/o, plain solution; w, solution with epinephrine (1:200,000) stippled bars. Compiled from Tucker GT, Moore DC, Bridenbaugh PO, et al. Systemic absorption of mepivacaine in commonly used regional block procedures. Anesthesiology 1972;37:277. |
Local Anesthetic Blood Concentrations and Toxicity
Commonly accepted thresholds for “CNS toxic” plasma concentrations from local anesthetic absorbed after uncomplicated
perineural administration range from 5 to 10 μg/mL for lidocaine, mepivacaine, and prilocaine, and from 2 to 4 μg/mL for bupivacaine, ropivacaine, and levobupivacaine. Higher values have a higher probability of associated toxic effects, and vice versa.
perineural administration range from 5 to 10 μg/mL for lidocaine, mepivacaine, and prilocaine, and from 2 to 4 μg/mL for bupivacaine, ropivacaine, and levobupivacaine. Higher values have a higher probability of associated toxic effects, and vice versa.
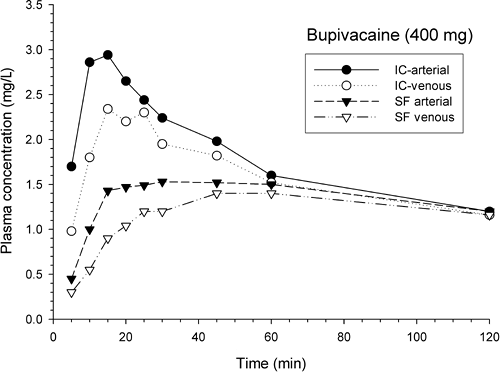 Figure 3-7. Arterial and venous mean plasma concentrations of bupivacaine (achiral assay) in surgical patients after intercostal or sciatic/femoral block procedures showing the magnitude and time course of the arteriovenous difference when sampled across the forearm. Compiled and redrawn from Moore DC, Mather LE, Bridenbaugh LD, et al. Arterial and venous plasma levels of bupivacaine (Marcaine) following epidural and intercostal nerve blocks. Anessthesiology 1976;45:39; and Moore DC, Mather LE, Bridenbaugh PO, et al. Arterial and venous plasma levels of bupivacaaine following peripheral nerve blocks. Anesth Analg 1976;55:763. |
It is intuitive that a greater circulating blood concentration of any local anesthetic agent is more likely to be associated with a greater risk of systemic toxicity than a lesser one. However, a deterministic relationship between blood drug concentration and toxicity is tenuous for several reasons. First, the rate of change of plasma drug concentrations is an important, but often neglected, element in the relationship because it relates to the degree of equilibration between tissue and blood drug concentrations—that is, how well the blood drug concentrations provide a reliable surrogate for the relevant tissue drug concentrations. Second, toxicity normally occurs concomitantly in different regions, and there can be both direct and indirect effects, for example, with cardiac effects deriving from direct myocardial effects and from CNS cardiac control mechanisms. Third, the concurrent physiologic and pharmacologic condition of the subject (e.g., blood gas/electrolyte/acid–base status, state of consciousness, etc.) can have marked influence on the outcome (148,151,152,166,167).
Several scenarios need to be distinguished pharmacokinetically: unusually rapid or normal persistent drug absorption from successful perineural administration, or unintended intravascular (usually IV, but occasionally intra-arterial) administration. Systemic toxicity from rapid absorption is usually acute, but relatively short-lived, whereas that from persistent absorption develops more slowly and recedes more slowly because the greater mass of drug deposited in the vital tissues takes longer to dissipate. Intravenous administration of sufficient dose usually produces sudden-onset, serious, acute generalized (whole body) effects that are usually rapidly receding, with a duration and severity related to the administered dose. Although reported relatively rarely, severely toxic effects can result from unintended intra-arterial injection of very small doses of local anesthetic agent into the afferent vasculature of vital organs (153,154), and this has been exploited with intended and appropriate doses as a useful research technique for studying regionally selective toxicity (155,156). Thus, it is important to remember that direct toxic effects in one region (notably, the brain) can have profound effects in another region (notably, the heart) via neural and cardiovascular system (CVS) mechanisms. Crucially, both the toxic effects and the associated blood drug concentrations depend on the state of consciousness, so that extrapolation of data between anesthetized and conscious subjects is especially hazardous (473,473a).
Most data documenting local anesthetic toxicity in humans are gathered, essentially opportunistically and retrospectively, from acute incidents in patients undergoing neural blockade, or from prospective studies in a small cohort of healthy volunteers undergoing administration of the drugs for experimental purposes. For ethical reasons, human subjects can be given only mildly toxic doses when local anesthetics are administered intravenously for research, typically to the threshold of CNS subjective symptoms (152,157,158,159,160,161,162). Information about more serious toxicity must, therefore, be derived either from clinical circumstances in which the objectives are preservation of life and well-being, rather than acquisition of scientific data, or from laboratory animal models. Laboratory rodent and isolated-tissue models are widely used and are especially useful for elucidating mechanisms and for comparisons between drugs (163,164,165); however, these are generally limited by the restricted data that can be obtained, or because of their destructive nature, or because of the isolation of tissues from their normal milieu. On the other hand, large experimental animals (dogs, pigs, sheep) can be prepared to allow pharmacokinetics and pharmacodynamics to be observed concurrently in a context rather similar to patient treatment. The ultimate goals of such experiments are normally to set “fail-safe” guidelines for human drug use on the grounds that responses among mammalian species are fundamentally the same, and to elucidate principles that can be expected to apply to humans using methods, techniques, and drug doses that are not normally offered in humans.
Table 3-4 Studies of threshold central nervous system toxicity in healthy young adult volunteer subjects performed with intravenous infusion of local anesthetic to a maximum dose or onset of subjective symptoms | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Although reported values such as those in Table 3-4 provide some useful guidelines, they refer to the mythic “average subject” and have to be interpreted in the light of many factors. These include whether measurements are made of plasma, serum, or blood drug concentrations; total or unbound (free) drug concentrations; relative enantiomer concentrations (if a racemic local anesthetic); active drug metabolite concentrations, as well as how the drug got into the plasma (intravenously or by vascular absorption), the rate of drug administration, the site of blood sampling (arterial or venous), how soon after drug administration the measured samples were drawn, and, most importantly, the physiologic status of the patient and, in particular, whether the patient was conscious and/or premedicated at the time (473a). In a previous era, the specificity and sensitivity of the drug assay procedure also could have been a factor, but this is rarely a concern with contemporary techniques. Thus, the values in Table 3-4 are more useful when circulating concentrations are not changing rapidly and there is time for equilibration between drug concentrations in plasma and vital tissues. Some individual subjects demonstrate toxic symptoms at lower drug concentrations than the commonly accepted values, whereas others do not demonstrate toxic symptoms despite having values within, or even greater than, those values (473b). In some cases, this is because of the development of locally high drug blood concentration. For example, local anesthetic drugs injected into patients with intracardiac right-left shunts (169) or injected inadvertently into the carotid or vertebral artery during attempted stellate ganglion block, bypass the lung, resulting in a high probability of CNS toxicity (153,154).
Extent and Rate of Absorption
In the absence of local metabolism, all of a dose deposited perineurally will eventually become absorbed into the systemic circulation. The concentration gradient is the main driving force for dissipation of drug both from its (heterogeneous) perineural site of administration into local tissues and uptake into the blood. The net resulting rate of systemic drug absorption will, therefore, be approximated by the sum of exponentialsf representing different rates of absorption from different local tissues, with the overall rate being governed by the agent’s distribution coefficient into, and the local perfusion of, the dominant tissue(s). A similar pattern pertains to drugs in the systemic circulation. Concentration gradients drive the exchange of drug between blood (strictly, plasma water) and tissues (strictly, extracellular fluids), including regions that excrete and metabolize drug. This pattern can also usually be represented by the sum of exponentials, in which the drug concentration approaches zero as time increases because of its excretion and/or metabolism lumped together as elimination (see systemic disposition, which is discussed later in the text).
If blood drug concentration–time profiles are available after perineural and IV administration, then it becomes possible to calculate the rate(s) of drug absorption. The underlying principle of this is that an IV injection reflects the whole-body (systemic) disposition of the drug; that is, distribution and elimination after it has arrived in the systemic circulation. Conversely, a perineural administration reflects the concurrent whole-body disposition of drug while arriving and once in the systemic circulation. The mathematical procedure of numerical deconvolution is used to determine the rate of leaving the (perineural) site of administration; this is depicted in Figure 3-8, where “pools” are shown to represent functional regions of drug deposition (rather than precise anatomic spaces). These calculations were originally performed for local anesthetics after IV regional anesthesia (170); subsequently, the method was extended to epidural anesthesia, with determination of the IV pharmacokinetics on a separate occasion (171). More recent technical developments have allowed application to a variety of subjects undergoing epidural and subarachnoid blocks, in which a concurrent small dose of deuterium-labeled agent is administered intravenously (172,173,174,175,176,177). The two forms of the
agent (native and labeled) are then separated analytically by mass spectrometry, and the rate of absorption of the native form used for the block is again determined by numerical deconvolution.
agent (native and labeled) are then separated analytically by mass spectrometry, and the rate of absorption of the native form used for the block is again determined by numerical deconvolution.
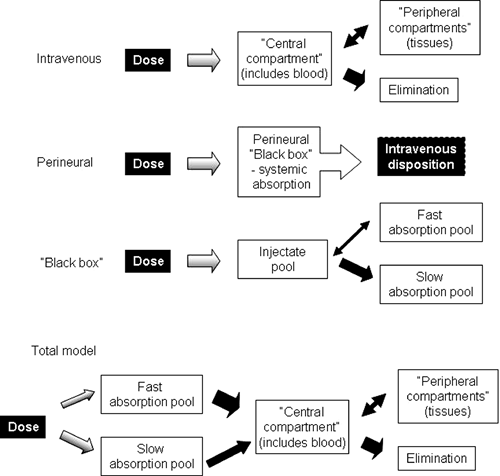 Figure 3-8. Conceptual model of local anesthetics injected perineurally used to calculate the rate of systemic absorption using the numerical deconvolution technique. The important assumption is that the systemic disposition of the drug, as revealed by intravenous administration, remains the same when the drug is administered perineurally. With the stable isotope method, the intravenous disposition is determined concurrently with the neural blockade. |
Such pharmacokinetic calculations provide a mathematical-statistical description of a functional process whereby the drug acts a tracer; nevertheless, the description has physical elements. Because local anesthetics are relatively lipophilic compounds they will partition into tissues around their site of administration. Thus, their vascular absorption rates will be directly related to blood flow and inversely to local tissue binding (distribution coefficient). Important local determinants of systemic absorption include those affecting the tissue distribution, including the site of injection, the lipophilicity and vasoactivity of the agent, the presence of additives such as vasoconstrictors, other formulation factors intended to modify local drug residence and release, the influence of nerve block, and (patho)physiologic features of the patient.
It has been found that systemic absorption after epidural administration can be described, in its simplest form, by a bi-exponential function; that is, the drug behaves as if it has been deposited into two portions with different absorption rate constants, roughly reflecting regions with identifiably different low-affinity/high-perfusion and high-affinity/low-perfusion characteristics. Although a mathematical construct, this can be likened to the drug physically partitioning between aqueous (faster absorption) and fatty (slower absorption) pools; the fraction of the dose going to the slower absorption pool increases with the more lipophilic agents (Table 3-5; Fig. 3-9) and/or when a vasoconstrictor is used (171,172,178). Tmax for a particular type of nerve block tends to occur at about the same time because the early absorption is dominated by the faster absorption rate constant, which is similar for all agents. However, the half-life of the drug in plasma is dominated by the slower absorption rate constant, and this is normally slower than the elimination rate constant. The real anatomic situation is, however, much more complex than a simple two-pool model.
Overall, for a given blood sampling site, Cmax and Tmax are inversely related in that the greater the Tmax then the lower the value of Cmax. Extensive data on Cmax and Tmax of the amide local anesthetics after various routes of injection have been tabulated elsewhere (145,178). So too have data on (poly)exponential equation exponents and coefficients describing the rate(s) of systemic absorption after epidural administration (171,172,173,174,175,176,177,178). Such data are mainly useful for comparison between drugs, or recipients of drugs.
Agent
Differences between agents need to be assessed under comparable conditions regarding drug administration, subject choice, and subject preparation. Inspection of Cmax and Tmax after epidural injection of plain solutions of the amide local anesthetics, and allowing for differences in sampling regimens, shows that the increment in peak whole blood drug concentration per 100 mg of dose is about 0.9 to 1.0 μg/mL for lidocaine and mepivacaine, slightly less for prilocaine, and approximately 50% to 60% as much for bupivacaine, etidocaine, ropivacaine, and levobupivacaine (167,172,178,179,180). Although differences in disposition kinetics contribute to this order, it appears that, despite similar times of peak concentrations, net absorption of the longer-acting, more lipophilic agents is slower. This is consistent with experimental data on residual concentrations of
the agents in epidural fat after injection into sheep (178) and is confirmed by deconvolution calculations of the time course of drug absorption in humans (173,174,175,178,180).
the agents in epidural fat after injection into sheep (178) and is confirmed by deconvolution calculations of the time course of drug absorption in humans (173,174,175,178,180).
Table 3-5 Mean (±SD) fractions and half-lives characterizing the absorption of lidocaine, bupivacaine, levobupivacaine, and ropivacaine after epidural and lidocaine and bupivacaine after subarachnoid administrations | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Differences in the net absorption rates of the various agents have implications for their accumulation during repeated and continuous administration. Whereas systemic accumulation is more marked with the short-acting amides, extensive local accumulation is predicted for the longer-acting compounds, despite their longer dosage intervals (171,181,182). Observations of relatively low blood concentrations of prilocaine with respect to the toxic threshold, particularly after brachial plexus block (Fig. 3-10) and IV regional anesthesia, support the claim that this compound should be the agent of choice for such single-dose procedures (183). For prilocaine and for articaine, a high systemic clearance, rather than slow absorption, is responsible mainly for the relatively low blood drug concentrations (12,183).
Although the rate of systemic absorption of local anesthetics is controlled largely by the extent of local binding, their relative intrinsic vasoactive properties could also modulate local perfusion and hence uptake. However, the effects are a complex function of drug, dose, enantiomer, and type and tone of blood vessel, and their relevance to the overall relative absorption of drugs after peripheral and central nerve blocks is difficult to evaluate (172). Nevertheless, it has been suggested, for example, that an increase in epidural blood flow (whether mediated locally or by change in cardiac output or both), with assumed increase in the systemic absorption rate of bupivacaine, is an important factor leading to regression of analgesia during the continuous epidural infusion of bupivacaine (184). Furthermore, the vasodilatory effect of bupivacaine on cerebral pial, cutaneous, and epidural blood vessels contrasts strikingly with the relative net vasoconstrictor effect of ropivacaine and levobupivacaine, a difference that may contribute to their relative anesthetic profiles (185–187a). Others, however, have found the effects on cutaneous blood vessels to be biphasic: vasodilatory at high concentrations and vasoconstrictive at low concentrations—clearly, the choice of exploratory model and conditions is critical in evaluating the net effects (188,189). Although R-bupivacaine was found to be two to three times more potent than S-bupivacaine in blocking nerve fibers in vitro (190), this intrinsic difference in action may be modulated in vivo by the dominant stereoselective effects on blood vessels. Thus, the S-enantiomer amide local anesthetics are longer acting than the R-enantiomers after separate subcutaneous injection, apparently reflecting a greater vasoconstrictor activity and, therefore, a presumed slower systemic absorption (14,191). Similar logic about the greater vasoconstrictor activity of ropivacaine than bupivacaine has been used to rationalize the slower apparent rate of ropivacaine absorption. For example, after caudal administration of 2 mg/kg doses of 0.2% solutions in children undergoing hypospadias repair, there was no significant difference in mean Cmax of the two agents, but the mean Tmax of ropivacaine occurred significantly later (65 minutes, c.f. 20 minutes). As the plasma concentrations were measured in peripheral venous blood, it is not possible to confirm this explanation because the Cmax Tmax values depended upon the rate of equilibration of drug between blood and
forearm tissues, and was complicated further by the patients being under isoflurane anesthesia (192).
forearm tissues, and was complicated further by the patients being under isoflurane anesthesia (192).
 Figure 3-9. Display of fraction of dose absorbed into the systemic circulation plotted as a function of time after epidural injection of selected amide local anesthetics, as determined by the stable isotope method in comparable patient populations. The curves represent bi-exponential functions fitted to the rate of systemic absorption. Compiled and redrawn from Burm, AGL, Vermeulen NPE, Van Kleef JW, et al. Pharmacokinetics of lidocaine and bupivacaine in surgical patients following epidural administration. Simultaneous investigation of absorption and disposition kinetics using stable isotopes. Clin Pharmacokinet 1987;13:191; Simon MJ, Veering BT, Stienstra R, et al. Effect of age on the clinical profile and systemic absorption and disposition of levobupivacaine after epidural administration. Br J Anaesth 2004;93(4):512–520; Simon MJ, Veering BT, Vletter AA, et al. The effect of age on the systemic absorption and systemic disposition of ropivacaine after epidural administration. Anesth Analg 2006;102:276–282; Veering BT, Burm AG, Vletter AA, et al. The effect of age on the systemic absorption, disposition and pharmacodynamics of bupivacaine after epidural administration. Clin Pharmacokinet 1992;22:75; Veering BT, Burm, AGL, Vletter AA, et al. The effect of age on systemic absorption and systemic disposition of bupivacaine after subarachnoid administration. Anesthesiology 1991;74:250; and Simon MJ, Veering BT, Stienstra R, et al. The systemic absorption and disposition of levobupivacaine 0.5% after epidural administration in surgical patients: A stable-isotope study. Eur J Anaesth 2004;24: 460–470. |
Site of Administration
Anatomic features, such as vascularity and the presence of tissue and fat that can bind local anesthetics, are primary influences on their rate of distribution within, and removal from, specific sites of injection. As revealed by imaging or dyes at various sites of local anesthetic injection, other features, such as the presence of septa or other fibrous tissues (and any resultant pressure gradients), also contribute to distribution of solution and thus spread of anesthesia (131,193,194,195,196). This is pertinent to all procedures of neural blockade, but particularly so for the central neuraxis, where the detailed anatomy is complex and obscured from the anesthesiologist, variable between individuals, and still largely unresolved with respect to the resultant block (197,198).
On the whole, and under comparable circumstances, as judged by Cmax values with regard for the effective dose, the net absorption rate decreases independently of the local anesthetic agent used, in the order intercostal block >caudal block >epidural block >brachial plexus block >sciatic and femoral nerve block (Fig. 3-6) (148,149,199,200). As indicated earlier, arterial plasma drug concentrations precede and exceed those in peripheral venous plasma, regardless of the agent. A study with epidural ropivacaine found that the forearm arteriovenous gradient diminished exponentially, with the difference essentially extinguished about 1 hour after administration (201), as would be expected from the time needed for drug concentration in the tissues to equilibrate with that in the venous blood. However, different conclusions might have been reached had the same study been performed by determining the drug arteriovenous concentration gradient in a lower limb because the block itself would have influenced the venous tone of the upper and lower limbs differently (171). Concurrent administration of general anesthetics and some medications can alter the time courses of arterial and venous plasma drug concentrations through local and systemic cardiovascular effects, and this may be observed when data from surgical patients and nonsurgical patients or healthy volunteers are compared.
Studies of epidural administration, as discussed earlier, found that the systemic absorption of several local anesthetics could be described as a bi-exponential process, differing only in the net rates; a similar description was proposed for paravertebral ropivacaine (192), and a mono-exponential process was described for subarachnoid lidocaine (172). These descriptions are consistent with anatomic findings, as demonstrated in sheep, of prolonged sequestration of more lipid-soluble local
anesthetic drugs in epidural fat. Thus, it is clear that the net rates of absorption are mainly sensitive to the lipophilicity of the agent and the fattiness of the region where the agents are deposited.
anesthetic drugs in epidural fat. Thus, it is clear that the net rates of absorption are mainly sensitive to the lipophilicity of the agent and the fattiness of the region where the agents are deposited.
 Figure 3-10. Plasma concentrations of local anesthetic agents after interscalene brachial plexus block (30 mL volume) with or without added epinephrine. Data from Wildsmith JAW, Tucker GT, Cooper S, et al. Plasma concentrations of local anaesthetics after interscalene brachial plexus block. Br J Anaesth 1977;49:237. |
Intercostal and Interpleural Blocks
The distribution of marker substances after injection into the costal grooves of cadavers has been investigated. It has been pointed out that variations in the anatomy of this region are far more common than has been commonly believed (202) and that information about the spread of solutions derived from cadaver studies may be misleading because of changes occurring postmortem (128). From studies of bupivacaine mixed with methylene blue dye in patients undergoing thoracotomy, Moorthy, et al. (128) concluded that the spread of anesthesia depends on the volume injected: a 5-mL injection spread through one intercostal space, but a 10-mL injection was more likely to spread to two or more intercostal spaces. Maximum absorbed concentrations of the agents after intercostal blocks using plain solutions are usually obtained rapidly, between 5 and 20 minutes, and may exceed individually toxic thresholds, but adverse systemic effects in patients, presumably, may be obtunded by the light general anesthesia/sedative premedication frequently used in patients undergoing these procedures. Since sustained high plasma drug concentrations are achieved during continuous intercostal infusions, supplementary bolus injections are likely to be dangerous (203).
Wide spreading of injectate occurs following interpleural space injection, with pooling in the paravertebral space (204,205). Exposure of drug to a relatively large surface area of tissue, resulting in rapid systemic absorption, emphasizes the potentially small safety margin of the interpleural block technique. Maximum plasma concentrations of bupivacaine are higher but occur later than when the same dose is injected for intercostal block (206). Dosage and plasma drug concentrations measured after interpleural injection of bupivacaine vary widely among studies. However, in patients undergoing cholecystectomy or laparotomy, van Kleef et al. (207) advocate the use of an initial bolus dose of 100 mg followed by continuous infusion of 5 mL/h of 0.25% bupivacaine with epinephrine. This has been shown to be safe, with no further gain in analgesic effect occurring with increased dose. Although Laurito et al. (208) observed that plasma bupivacaine concentrations often increased above the putative toxic threshold of 2 μg/mL using the same infusion rate as van Kleef et al. (206), no adverse effects were observed; this may reflect the postoperative rise in plasma drug binding. In contrast to van Kleef et al. (207) and Laurito et al. (208), who measured continually rising plasma drug concentrations throughout prolonged infusion, Kastrissios et al. (209) observed a steady state.
Epidural and Caudal Blocks
Local anesthetic solution injected epidurally spreads cephalad and caudad as if in a distensible, but leaky, sleeve in which the local anesthetic solution concurrently leaks from foraminae and the local anesthetic agent diffuses into tissues, including dorsal root ganglion cells, and into the CSF (67,71,72,197,198). Although the relationship between injected volume (discrete dose) and number of anesthetized dermatomes has been described mathematically by a cubic polynomial equation (210), this has no physical significance. Not surprisingly, individual variability in the surface area and composition of the epidural space are primary determinants of epidural anesthesia (211,212).
The results of studies on the spread of analgesia indicate a minor influence of injection speed (213,214,215). It has been suggested that, for comparisons, the dose/segment is a more useful outcome variable than spread alone (216,217). Confounding factors include the drug used, the age and size of the patient, the range of injection pressures, and the approach and direction of the needle bevel (175,196,218,219,219a). It has been claimed that the presence or absence of epinephrine is not a significant factor (216). Insofar as epidural opioids can provide analogies for local anesthetics with similar physicochemical properties, the effects of epinephrine on spinal pharmacokinetics has been found to be more complex than can be explained by simple “greater local retention,” and is not predictable from simple observation of plasma drug concentrations (132).
Overall, after epidural administration, the extent of local anesthesia results from cephalad–caudad spread of solution and diffusion of local anesthetic to the spinal cord and nerve roots, as well as transport within the CSF. The apparent rates of systemic absorption and fractions of the dose transferred into CSF of the local anesthetics are inversely proportional to lipophilicity (67). This suggests that competition for drug between the various distribution and clearance processes after epidural administration favors vascular uptake (irreversible local clearance) over transmeningeal distribution. Nevertheless, local anesthetics appear rapidly in the CSF after epidural injection (72,220). Peak drug concentrations in the CSF occur within 10 to 20 minutes and are sufficient to produce blockade
of spinal nerve roots. By 30 minutes, high drug concentrations are also achieved in the peripheral cord and in the spinal nerves in the paravertebral space (126). Apart from direct diffusion of drug across the dura, access to the cord, particularly the dorsal horn region, may be mediated (a) by diffusion; (b) by uptake into the posterior radicular branch of spinal segmental arteries, although subsequent work has suggested that this might be more a source of clearance from the cord rather than uptake (70,117); (c) by centripetal subneural and subpial spread from the remote paravertebral nerve trunks (126); and (d) by bulk flow through the arachnoid via the dural root sleeves, although the latter has not been supported by studies using dura ex vivo (70). These suggestions are consistent with clinical observations of the distribution of analgesia during induction and regression of epidural block. A segmental pattern of analgesia during onset may be related to the initial drug diffusion into spinal nerves and roots, with subsequent nonsegmental regression resulting from ultimate diffusion to structures within the cord (221). It is also clear that even a very small hole made during the introduction of an epidural needle can give rise to spuriously high “rates of diffusion” of drugs injected epidurally into the subarachnoid space (222).
of spinal nerve roots. By 30 minutes, high drug concentrations are also achieved in the peripheral cord and in the spinal nerves in the paravertebral space (126). Apart from direct diffusion of drug across the dura, access to the cord, particularly the dorsal horn region, may be mediated (a) by diffusion; (b) by uptake into the posterior radicular branch of spinal segmental arteries, although subsequent work has suggested that this might be more a source of clearance from the cord rather than uptake (70,117); (c) by centripetal subneural and subpial spread from the remote paravertebral nerve trunks (126); and (d) by bulk flow through the arachnoid via the dural root sleeves, although the latter has not been supported by studies using dura ex vivo (70). These suggestions are consistent with clinical observations of the distribution of analgesia during induction and regression of epidural block. A segmental pattern of analgesia during onset may be related to the initial drug diffusion into spinal nerves and roots, with subsequent nonsegmental regression resulting from ultimate diffusion to structures within the cord (221). It is also clear that even a very small hole made during the introduction of an epidural needle can give rise to spuriously high “rates of diffusion” of drugs injected epidurally into the subarachnoid space (222).
Most studies have found no relationship between the various indices of body size and plasma concentrations of local anesthetic agents, but Sharrock et al. suggested that 20% to 40% of the variance in arterial plasma concentrations of bupivacaine could be explained by correlation with body surface in elderly surgical patients (223). The effect of fat deposits within the epidural space in delaying the absorption of local anesthetics has been discussed earlier, and this is the likely explanation for these observations. Local anesthetic will be taken up into the extradural veins and pass from there to the azygos vein. In the presence of raised intrathoracic pressure, absorbed drug could also be redirected up the internal vertebral venous system to cerebral sinuses. Vascular absorption of local anesthetic from different regions of the epidural space (cervical, lumbar, thoracic) appears to be similar (224), although the cardiovascular sequelae may differ markedly and thereby affect the systemic disposition of the agent.
Epidural infusions of local anesthetics are now in widespread use (225). When administered by infusion, the plasma concentrations will increase if the administration rate exceeds the total body clearance. Bupivacaine and ropivacaine both produce good pain relief in obstetric patients, but the plasma concentrations of ropivacaine were found to be greater than those of bupivacaine when administered at equal rates, despite the former having a greater mean total body clearance; such a discrepancy is, therefore, likely to be the result of the net slower absorption of bupivacaine (226). Others have found that ropivacaine plasma concentrations increased with duration of infusion in patients after surgery and were proportional to dose rate, indicating that the administration rate exceeded the mean total body clearance at that time (225a,225b,225c, 335,342,525,530,533).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


