Key Clinical Questions
What are the causes of potassium and magnesium disorders?
What are the potential consequences of potassium and magnesium disorders?
How are potassium and magnesium disorders treated?
How are potassium disorders treated in clinical situations with rapid potassium shifts, such as diabetic ketoacidosis, hyperglycemic hyperosmolar state, and periodic paralysis?
Introduction
Potassium and magnesium disorders are common in the hospital setting. About 12% of hospitalized patients have hypokalemia, 3% have hyperkalemia, and 11% have hypomagnesemia. Potassium disorders are a particular challenge for hospitalists, as the first clinical manifestation of a severe potassium abnormality may be a cardiac arrhythmia.
Potassium (K+) is the most abundant intracellular cation. In a 70 kg adult, the total body K+ is approximately 3500 mmol (50 mmol/kg). About 98% is located in the intracellular compartment, with the majority localized in muscle (70%). Because only 2% of the total body K+ is in the extracellular compartment, laboratory results correlate poorly with total body K+. Normal levels of extracellular potassium are between 3.5 and 5.0 mmol/L, while intracellular K+ levels range between 140 and 150 mmol/L.
Similarly, plasma magnesium (Mg2+) accounts for only 1% of the total body magnesium stores, and plasma Mg2+ correlates poorly with total body magnesium. Plasma magnesium is normally 1.7 to 2.3 mg/dL (0.70–0.95 mmol/L). Magnesium abnormalities can also have major clinical consequences, such as torsades de pointes. Hypokalemia is often accompanied by hypomagnesemia, which renders potassium repletion less effective.
Potassium Balance
Dietary K+ is excreted mostly in the urine (90%), with a minor component of fecal excretion (10%). In severe chronic kidney disease, stool losses may exceed 25% to 50% of dietary intake. Under normal conditions, the kidney can adjust potassium excretion over a wide range depending on K+ intake. The recommended daily K+ for patients with normal renal function is 4700 mg (120 mmol). If K+ control mechanisms are intact, an increased daily intake of 400 mmol can be tolerated with < 1 mmol/L increase in plasma K+ values. Daily K+ intake for patients on hemodialysis or with severe chronic kidney disease is typically 2000 mg (51 mmol) per day. Conversely, many patients on peritoneal dialysis can tolerate a normal daily intake of K+ due to losses of potassium in the urine and peritoneal fluid.
Potassium unit conversion
|
Hyperkalemia
Hyperkalemia is defined as a plasma K+ > 5.0 mmol/L. Hyperkalemia can result from redistribution, reduced K+ excretion, or increased K+ intake. Medications may lead to hyperkalemia by affecting either potassium redistribution or excretion (Table 250-1).
| Mechanism | Medications |
|---|---|
| Increased potassium load | KCL, penicillin G |
| Renin release inhibition | Beta-blockers, NSAIDs, COX-2 inhibitors |
| Angiotensin I to II conversion inhibition | ACE inhibitors |
| Angiotensin II receptor blockade | ARBs |
| Aldosterone synthesis inhibition | Heparin |
| Aldosterone receptor blockade | Spironolactone, eplerenone |
| Renal epithelial Na channel; (ENaC) blockade | Triamterene, amiloride, Trimethoprim, pentamidine |
| Inhibition of Na-K-ATPase | Digoxin, calcineurin inhibitors (cyclosporin, tacrolimus) |
| Drug-induced rhabdomyolysis | Statins, cocaine |
| Drug-induced tumor lysis | Selected chemotherapy agents |
| Transcellular shift | Succinylcholine |
Pseudohyperkalemia is a laboratory artifact due to potassium release from cells prior to laboratory measurement. It should be suspected when hyperkalemia is reported in the setting of red blood cell hemolysis, leukocytosis (> 70,000/mm3) or thrombocytosis (> 500,000/mm3), and no ECG changes are present. It has become less common with the increased use of plasma for potassium measurement, rather than serum. Plasma is the supernatant collected from heparinized whole blood, whereas serum is the supernatant remaining after centrifugation of a clotted whole blood sample. Serum K+ is normally 0.3 mmol/L above the plasma K+, but may be higher if K+ is released from the clot formed in the tube. If pseudohyperkalemia is suspected, the plasma potassium should be measured instead of the serum potassium. Pseudohyperkalemia can also occur due to potassium release from muscle cells following prolonged constriction with a tourniquet or limb exercise while a tourniquet is in place.
Redistribution of potassium from the intracellular to the extracellular space can occur in severe acidosis due to nonorganic acid metabolic acidosis, hyperosmolar states, tissue breakdown, and hyperkalemic periodic paralysis. Organic acids such as lactic acid and ketoacids are less likely to cause hyperkalemia than non-organic acids. These organic acids have greater transmembrane mobility, allowing movement into cells with H+, rather than movement of K+ out of cells in exchange for H+. Hyperkalemia in diabetic ketoacidosis usually results from insulin deficiency and hyperosmolality, rather than acidosis. Hyperglycemia increases extracellular osmolality, drawing water from cells down the osmotic gradient. Potassium follows the water movement (solvent drag), and hyperkalemia results.
Tissue breakdown may liberate large amounts of intracellular potassium, resulting in rapid, life-threatening increases in extracellular potassium. Rhabdomyolysis, tissue necrosis, tumor lysis with chemotherapy, and large hematomas are common causes. For rhabdomyolysis, hypokalemia may precede hyperkalemia because of vasoconstriction and decreased blood flow to the involved muscle.
Hyperkalemic periodic paralysis is an autosomal dominant disorder involving the muscle cell sodium channel. During these episodes, potassium moves from the intracellular to extracellular space, accompanied by movement of sodium and water into the cell. The hyperkalemia is accompanied by weakness or paralysis.
Potassium excretion in the distal nephron depends upon adequate urine flow, aldosterone, and activity of the basolateral Na+/K+-ATPase. The reabsorption of luminal Na+ through aldosterone-sensitive epithelial sodium channels (ENaCs) creates an electrochemical gradient for K+ excretion in the urine. Hyperkalemia can occur in aldosterone-resistant or deficient states by attenuating the electrochemical gradient for K+ secretion. Acquired mineralocorticoid resistance from diabetes mellitus, obstructive uropathy, chronic tubulointerstitial disease, sickle cell anemia, lupus nephritis, and medications, as well as decreased mineralocorticoid production (including medication induced), are commonly associated with a type IV renal tubular acidosis (RTA). Type IV RTA is characterized by hyperkalemia, normal anion gap hyperchloremic acidosis with serum HCO3 values ranging between 16 and 22 mmol/L, and decreased urinary NH4+ production, causing a positive urine anion gap [Urine (Na+) + (K+) – (Cl–)]. In acute and chronic tubular injury, a decreased responsiveness to aldosterone can contribute to hyperkalemia. For acute kidney injury associated with prerenal azotemia or hypovolemic states, decreased urinary flow to the distal tubule and a reduction in sodium-potassium exchange can contribute to hyperkalemia.
Excessive intake of K+ rarely causes hyperkalemia in the setting of normal renal function. Renal secretion of potassium is typically adequate at glomerular filtration rates (GFR) above 20–30 ml/min/1.73 m2. Patients with end-stage renal disease (ESRD) on hemodialysis usually tolerate a daily K+ intake of 2000 mg (51 mEq). Upregulated gut potassium excretion and shifts in transcellular K+ prevent hyperkalemia between dialysis sessions. Loss of these mechanisms may result in the rapid development of hyperkalemia, especially with large exogenous loads of potassium, such as massive blood transfusions. Irradiation of blood and increased age of the blood increase the amount of free potassium that is released during the blood transfusion. Seven-day-old blood has approximately 23 mmol/L of K+, while 42-day-old blood has approximately 50 mmol/L of K+.
|
Clinical effects of hyperkalemia relate to altered membrane excitability due to changes in the transcellular potassium gradient. Severe hyperkalemia leads to cardiac arrhythmias and conduction abnormalities. It may also cause weakness of the lower extremities, progressing superiorly to cause flaccid paralysis and respiratory failure. This presentation may mimic Guillain-Barré syndrome, but is easily differentiated by the response to potassium correction. Hyperkalemia may also contribute to metabolic acidosis by interfering with renal ammonium excretion.
In the hospital setting, the initial evaluation of hyperkalemia includes monitoring for life-threatening arrhythmias, checking for pseudohyperkalemia, eliminating exogenous sources of potassium, evaluating renal function, and evaluating for rapid transcellular shifts of potassium (Figure 250-1).
Figure 250-1
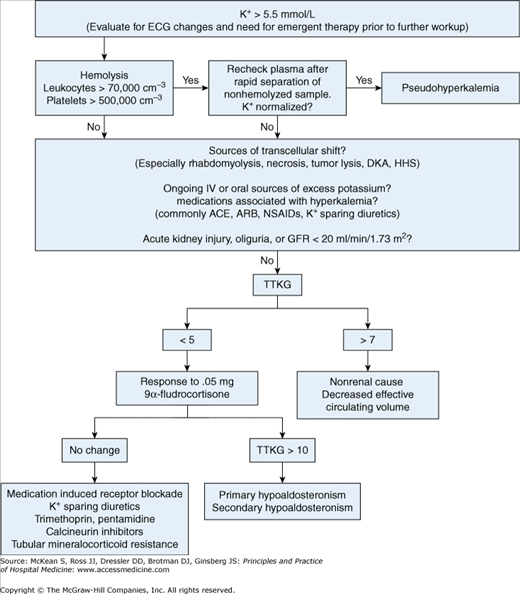
Diagnostic assessment of hyperkalemia. (ACE, angiotensin-converting enzyme inhibitor; ARB, angiotensin receptor blocker; DKA, diabetic ketoacidosis; ECG, electrocardiogram; GFR, glomerular filtration rate; HHS, hyperosmolar hyperglycemic state; NSAIDs, nonsteroidal anti-inflammatory drugs; TTKG, transtubular potassium gradient.)
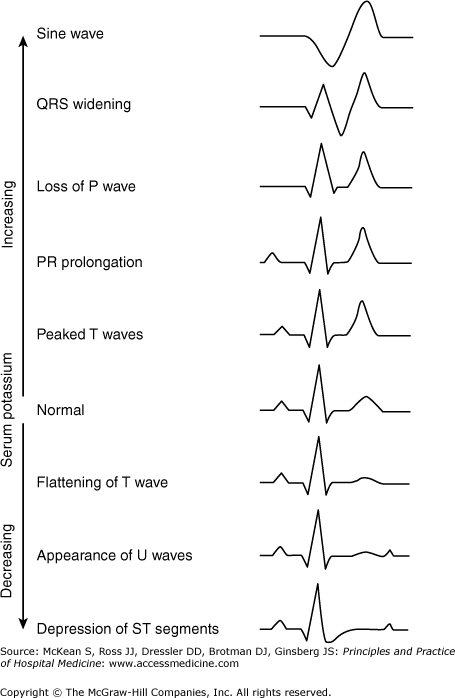
Even mildly elevated K+ levels may be associated with ECG changes, especially in the setting of rapid rises in plasma K+ values. If ECG changes from hyperkalemia are found or if the plasma K+ value is ≥ 7.0, continuous telemetry is indicated and should continue until the plasma K+ value is ≤ 5.8 and there are no hyperkalemic ECG changes. Frequent chemistry checks are indicated for plasma K+ greater than 5.8 or for hyperkalemia associated with ECG changes, severe hyperglycemia, or any clinical condition which predisposes the patient to rapid changes in plasma K+.
Plasma potassium levels do not correlate with specific ECG changes. A symmetric increase in T wave height may be seen initially. Hyperkalemia-induced peaked T waves may be difficult to distinguish from the hyperacute T waves of myocardial injury. As potassium levels rise further, flattening of the P waves, prolongation of the PR interval, and prolonged QRS duration occur. Eventually, atrial standstill and a sine wave ECG pattern may be seen (Figure 250-2).
A 24-hour urine K+ measurement can help differentiate between renal and nonrenal causes of hyperkalemia. With hyperkalemia, urinary excretion of K+ should exceed 40 mmol/d. Alternatively, the transtubular potassium gradient (TTKG), a measurement of potassium secretion by the distal nephron corrected for urine osmolality, can be used:
TTKG = (KU/KS) × (SOsm/UOsm)
KU and KS are the concentrations of K+ in the urine and serum, and SOsm and UOsm are the osmolalities of the serum and urine, respectively. Plasma potassium and osmolality can be used instead of serum values to estimate TTKG with minimal effect on clinical interpretation.
Patients with normal renal function and normal potassium intake have a TTKG of 8 to 9. In hyperkalemia, a low TTKG (< 5–7) suggests an inappropriately low secretion of potassium. A high TTKG with hyperkalemia suggests normal aldosterone action and an extrarenal cause of hyperkalemia, except in cases of volume depletion where aldosterone secretion is enhanced with a TTKG > 7, but total renal potassium excretion is limited by low urine flow.
Related posts:
 Strategies for Cost-Effective Care
Strategies for Cost-Effective Care
 Building, Growing, and Managing a Hospitalist Practice
Building, Growing, and Managing a Hospitalist Practice
 Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
Designing a Hospitalist Compensation and Bonus Plan
The Face of Health Care Emerging Issues for Hospitalists
 Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist
Medical Malpractice
Preventing and Managing Adverse Patient Events: Patient Safety and the Hospitalist
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


