Key Clinical Questions
How and when do you evaluate pituitary hormonal function?
How do you treat patients with excess or deficient pituitary hormonal function?
How do you classify pituitary tumors?
Which patients with pituitary disease require hospital admission?
What are the indications for consultation with endocrinology and/or neurosurgery?
What are the complications associated with neurosurgical resection of pituitary tumors?
Introduction
Pituitary disorders are characterized by tumors that lead to excessive hormone secretion or by inadequate hormone production from a multitude of causes. Pituitary disease is more common than popularly supposed. In unselected autopsies, the prevalence of pituitary adenomas averages about 14%. However, the prevalence of clinically significant pituitary adenomas is much lower, about 94 per 100,000 people. At least 30% of survivors of traumatic brain injury and subarachnoid hemorrhage have chronic anterior hypopituitarism at 24–36 months; therefore, clinicians must consider this diagnosis in populations at risk, such as wounded combat veterans. Hypopituitarism is also frequent after cranial irradiation, and may complicate pituitary disease of any cause. This chapter discusses the basic function of the pituitary gland, the evaluation of hypopituitarism, and the approach to patients with pituitary tumors.
Pituitary Gland Anatomy and Physiology
The pituitary gland is located at the base of the brain within the sella turcica, which is the bony roof of the sphenoid sinus. It lies outside the dura mater. The pituitary stalk, containing neurovascular bundles, extends from the hypothalamus through a dural opening to the pituitary gland. This anatomical arrangement renders the pituitary stalk vulnerable to traumatic injury. The optic chiasm is located above the pituitary gland and anterior to the pituitary stalk. The pituitary gland is bordered on the sides by the cavernous sinuses, which consist of vascular lakes containing cranial nerves III, IV, V, and VI and a portion of the carotid artery. The pituitary gland consists of two lobes, anterior and posterior (Figure 152-1).
Figure 152-1
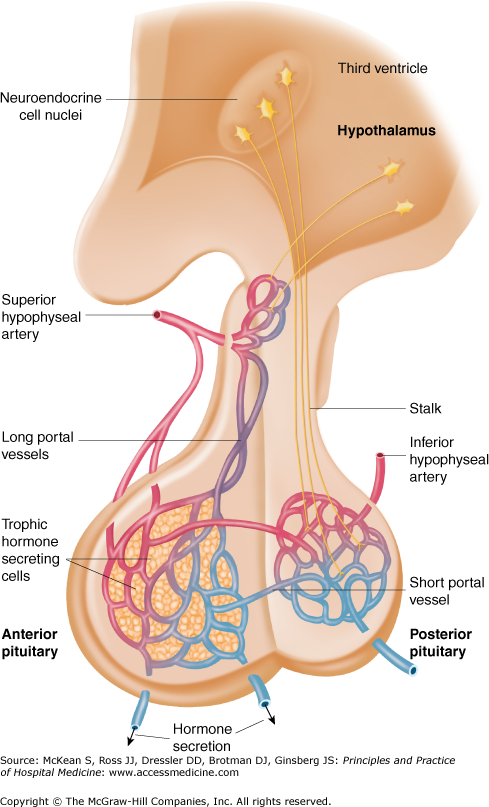
The hypothalamic-pituitary axis. Hypothalamic nuclei secrete hormones in a pulsatile fashion into a portal venous system, which perfuses anterior pituitary secretory cells and regulates its activity. Posterior pituitary hormones are derived from direct neural extensions from the hypothalamus. (Reproduced, with permission, from Fauci AS, Braunwald E, Kasper DL, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill, 2008. Figure 333-2.)
The anterior lobe, or adenohypophysis, is composed of secretory cells that manufacture at least six hormones: corticotropin (ACTH), thyroid stimulating hormone (TSH), growth hormone (GH), follicle stimulating hormone (FSH), luteinizing hormone (LH), and prolactin (PRL). Secretion of the first five hormones is triggered by hypothalamic releasing factors that travel down the pituitary stalk through a portal venous system. In contrast, anterior pituitary secretion of prolactin, which promotes lactation, is tonically inhibited by hypothalamic secretion of dopamine. Any disorder that compresses or interferes with the pituitary stalk can cause prolactin elevation.
In contrast, the posterior lobe, or neurohypophysis, is comprised of axons of cell bodies arising in the hypothalamus. These axons store and secrete vasopressin (antidiuretic hormone, or ADH) and oxytocin. Vasopressin has a key role in fluid and electrolyte balance by promoting retention of water by the kidney. Oxytocin stimulates uterine contractions at parturition.
Hypopituitarism
The most common cause of hypopituitarism is a pituitary adenoma. Other causes include nonpituitary sellar masses, such as craniopharyngiomas and meningiomas, damage to normal pituitary tissue during neurosurgery for pituitary adenoma, and sudden hemorrhage into a pituitary adenoma (pituitary apoplexy). Sheehan syndrome is pituitary necrosis related to postpartum hemorrhage and hypotension; presumably the pituitary is at greater risk of ischemic injury in this setting because of its normal increase in volume during pregnancy. Patients usually present with failure of lactation, followed by slowly progressive symptoms and signs of hypopituitarism. Cranial irradiation causes hypopituitarism in approximately 40% of patients over a 10-year period, with the precise risk depending on the total radiation dose and length of time after exposure. The prevalence of hypopituitarism after traumatic brain injury (TBI) and subarachnoid hemorrhage (SAH) has been reported to be approximately 30% and 50%, respectively. Growth hormone deficiency is the most common endocrine abnormality following both TBI and SAH. Developmental and genetic causes of hypopituitarism are rare, and include pituitary aplasia or hypoplasia, hypothalamic defects, mutations in transcription factors, and genetic conditions, such as Kallman syndrome and Prader-Willi syndrome. Other rare causes include metastatic disease from breast, lung, or skin cancer, histiocytosis X, and sarcoidosis.
Presentation and Diagnosis
| Pituitary Hormone | End Organ and its Hormone | Criteria for Hormonal Excess | Criteria for Hormonal Deficiency |
|---|---|---|---|
| ACTH | Adrenal glands: Cortisol |
|
|
| TSH | Thyroid gland: T4, T3 |
|
|
| Growth hormone (GH) | Liver: IGF-1 |
|
|
| FSH, LH | Testes: testosterone |
|
|
| Ovaries: estradiol, progesterone |
|
| |
| Prolactin | Mammary glands | ⇑ Prolactin | ⇓ Prolactin (rare) |
Central or secondary adrenal insufficiency, caused by decreased pituitary ACTH release, is associated with anorexia, orthostasis, arthralgias, fatigue, vomiting, and hyponatremia. Hyperpigmentation is absent and serum potassium is normal, unlike Addison disease (primary adrenal insufficiency). The evaluation of central adrenal insufficiency is somewhat controversial, as the criteria for diagnosis depend on the severity of the patient’s condition. In a stable, noncritically ill patient, a fasting serum cortisol > 15 μg/dl is considered normal. Central adrenal insufficiency is clearly present when the morning serum cortisol is < 3 μg/dl and ACTH is low or inappropriately normal. If the serum cortisol is between 3 and 15 μg/dl, then further testing is appropriate. Such testing may include a standard 250 μg ACTH (cosyntropin) stimulation test, in which serum cortisol is measured at baseline and 30 and 60 minutes after intravenous cosyntropin administration. Alternatively, serum cortisol may be measured 45–60 minutes after an intramuscular injection of cosyntropin. A peak serum cortisol of > 18 μg/dl is considered normal in this test. An important limitation of the use of cosyntropin stimulation testing in central adrenal insufficiency is that it may be normal in the acute setting of pituitary disease (such as following pituitary surgery) because the adrenal gland can respond normally to cosyntropin in the first few weeks following a pituitary insult. Over time, ACTH deficiency leads to adrenal atrophy, and at that point the response to cosyntropin stimulation is blunted. It is also possible that the pharmacologic dose of cosyntropin may overshadow mild central adrenal insufficiency, and a normal result may give a false sense of security that adrenal function is intact. Other measures of ACTH reserve, such as metyrapone stimulation or insulin-induced hypoglycemia testing, may be more reliable as they measure integrity of the entire axis, but they are also less practical and rarely used in clinical practice. The diagnosis of central adrenal insufficiency in the presence of physiologic stress is less clear, as a “normal” cortisol value may be inappropriately low for the setting. Change in serum cortisol following cosyntropin has been used as a marker for adrenal function in the intensive care unit, but there is controversy as to the interpretation of such tests.
The symptoms of central hypothyroidism are similar to those of primary hypothyroidism, and include cold intolerance, fatigue, mild weight gain, dry skin, alopecia, and constipation. Findings include delayed tendon reflexes, facial puffiness and coarsening, bradycardia, slowed mentation, nonpitting edema, pleural effusions, and carpal tunnel syndrome. Diagnosis is based on the presence of a low serum free T4 with low or normal TSH, in contrast to the elevated TSH seen in primary hypothyroidism. However, low free T4 can be the first thyroid abnormality associated with nonthyroidal illnesses (sick euthyroid), common in the inpatient setting. Therefore, the definitive diagnosis of central hypothyroidism may sometimes be delayed until the subject recovers from acute illness.
Hypogonadism (testosterone and estrogen deficiency) cannot be reliably diagnosed in the inpatient setting, as levels of both hormones decline markedly during acute illness. In men, secondary hypogonadism is associated with decreases in sexual function, libido, and muscle mass. In women, secondary hypogonadism is associated with secondary amenorrhea and symptoms of estrogen deficiency, such as hot flashes, vaginal dryness, or decreased libido. In the outpatient setting, secondary male hypogonadism is diagnosed based on low levels of testosterone and low or normal FSH and LH. In women, secondary hypogonadism is suggested by irregular or absent menses and low or normal FSH and LH.
Growth hormone (GH) deficiency in children is characterized by short stature, and in adults by increased body fat, reduced muscle mass and bone density, and diminished quality of life. GH is secreted in pulsatile fashion, and most secretion is nocturnal. During the day, GH levels in normal subjects may be low or undetectable. Therefore, random daytime GH levels are of little clinical utility. As GH stimulates production of insulin-like growth factor-1 (IGF-1) by the liver, IGF-1 is a surrogate marker of recent GH levels. A low serum IGF-1 level supports the diagnosis of GH deficiency, especially in the setting of other pituitary disease. Further evidence of GH deficiency may be provided by an insufficient GH response to insulin-induced hypoglycemia or to growth hormone-releasing hormone (GHRH).
Central diabetes insipidus (DI) due to ADH deficiency presents with polyuria and polydipsia. The physical examination is normal unless dehydration has developed. If hypernatremia is present, the patient may be obtunded, but most patients are able to maintain eunatremia with a high fluid intake. The diagnosis is confirmed with a carefully supervised fluid deprivation test, demonstrating the inability to appropriately concentrate urine osmolality despite rising plasma sodium and osmolality. Patients with central DI will subsequently increase their urine osmolality more than 50% with desmopressin administration, whereas patients with nephrogenic DI will not.
Pituitary Tumors
Pituitary tumors are classified based on hormonal secretion and size. Tumors that secrete excessive amounts of pituitary hormones are referred to as functional or secretory tumors, and tumors that do not produce biologically active hormone are known as nonfunctioning or nonsecretory tumors. Pituitary tumors are classified as microadenomas if the diameter is less than 10 mm, and macroadenomas if the diameter is greater than 10 mm.
Patients with functioning pituitary adenomas present with classic manifestations specific to each hormone (Table 152-2). These benign tumors usually secrete in order of decreasing incidence excessive quantities of prolactin, growth hormone, ACTH, and rarely thyrotropin, or gonadotropin.
|







