Pharmacologic Substrates of Pain: Peripheral Voltage Gated Ion Channels in Pain
Lucinda C. Seagrove
Anthony H. Dickenson
Peripheral nerves convey external sensory information to the spinal cord. At this first central level, a large amount of convergence and modulation occurs before transmission to the higher centers of the brain where the final perception of the stimulus is established. In addition, descending pathways from the brain to the spinal cord can further shape the output of the dorsal horn by way of inhibitory and facilitatory influences. Thus, processing occurs from both a “bottom-up” and a “top-down” series of events.
However, apart from the poorly understood processes of central pain, activity in peripheral sensory neurons is critical to the genesis of pain. The peripheral terminals of sensory neurons allow the transduction of various modalities such as mechanical, thermal, and chemical stimuli via a number of different proteins, receptors, and ion channels that convert the particular modality into electrical impulses. Critical to this are voltage-gated Na+ channels (VGSCs), which initiate and propagate action potentials that travel the length of the axons, promote neuronal excitability (Fig. 33-1), and lead to the release of transmitters, in the case of sensory neurons, glutamate and peptides. Release of these transmitters into the spinal cord activates receptors on spinal neurons that in turn leads to the activation of projection neurons and interneurons within spinal circuitry.
These mechanisms are fundamental to normal sensation, whether it is signaling innocuous everyday tactile or thermal stimuli or acute painful stimuli, in which case these sensations serve as a warning. However, in pathologic pain states, for example those resulting from peripheral nerve or tissue damage due to disease or trauma, these same mechanisms can become distorted, so that the system (a) becomes hyperexcitable and sensory transmission persists, (b) can be generally not reflective of the magnitude of the evoking stimulus, and (c) can start to generate spontaneous, stimulus-independent activity as well as abnormal evoked activity.
Preclinical experimental studies have been, and continue to be, invaluable in uncovering both physiologic and pathophysiologic mechanisms responsible for sensory signaling and chronic pain conditions, respectively. However it still remains a challenge to translate such findings to clinically observed symptoms and possible therapeutic strategies (1). Blockade of the major drive to activity in cells—the voltage-gated Na+ channel (VGSC)—is a major target in current clinical practice in several therapeutic areas including epilepsy, arrhythmias, and pain, and a number of studies have highlighted Na+ (Na+) channels as important mediators in neuropathic pain. Widely used local anaesthetics, such as lidocaine, act as Na+channel blockers, and this verifies the potential for targeting these channels as analgesic strategies. In neuropathic pain patients, microneurographic recordings from their peripheral nerves reveals aberrant electrical activity, which interestingly can be reduced by local anaesthetic nerve block in parallel with an attenuation in symptoms (2,3). Further support for the concept that Na+channels are valid targets comes from the evidence for a therapeutic effect of other drugs with VGSC blocking actions, namely carbamazepine, mexiletine, and lamotrigine (1,4).
Within the nervous system, at least nine subtypes, defined by their pore-forming α-subunit, each with individual functional and expressional characteristics, are expressed (5), with most dorsal root ganglion (DRG) neurons expressing all types (6). Voltage-gated Na+channels are not restricted to the central nervous system (CNS) and are also found in other excitable tissues, notably, the heart, autonomic neurons, and skeletal muscle. Therefore, without specificity of action or use-dependency, block of Na+channels per se runs the risk of complete sensory anaesthesia and intolerable side-effects due to potential ubiquitous block of cardiac and brain electrical activity. Although Na+ channel blocking drugs (local anaesthetics, anticonvulsants, antiarrhythmics) used clinically for pain and other indications exhibit some degree of use-dependency in that normal low frequency neuronal firing is not affected, whereas pathologic high-frequency patterns of activity are reduced (7,8), the therapeutic window remains low. Developments in regional blocks can circumvent some of these problems but more selective drugs would be helpful.
Despite many attempts to modify the existing agents and ongoing attempts to identify VGSC subtype-specific ligands, currently no specific agents exist. As a consequence, the roles and potential differential physiologic roles of individual Na+ channel subtypes cannot be studied in vivo by pharmacologic means. Fortunately, in animals, other methodologic approaches can be employed, such as genetic modifications to knock out specific Na+ channel subtypes, a practice which has shed considerable light on the roles of various Na+ channels, as has oligodinucleotide (ODN) antisense knock-down protocols, in which the target protein is temporarily reduced.
Progress in this therapeutic area is therefore dependent on translation of findings from animal studies. For this to be of any use, the animal models, methods of assessment, and inferences drawn need to be carefully established to link the animal biology to the human condition. However, a combination of molecular, genetic, behavioral, and electrophysiologic studies can be highly informative in this respect. The issue with animal
models is that subtle side effects of drugs can be next to impossible to gauge, and so the preclinical approaches will primarily inform on efficacy. Consideration is also needed on what is being measured in animals. A large majority of studies are behavioral, in which the response to a stimulus is defined by the time or force to withdrawal of the animal at a certain intensity of the stimulus. This will be close to the threshold for “pain.” Thus, a drug may have reasonable efficacy against a pain of this level but lose or even gain effectiveness at higher intensities, more related to the pains that give rise to clinical problems. This problem can be overcome by recording neuronal activity in vivo (although potentially complicated by general anaesthesia), in which responses of defined sensory neurons, for example in spinal cord, thalamus, or other areas with somatosensory inputs, can be quantified using stimuli that would be above the threshold. Effects on threshold and suprathreshold responses can therefore be revealed. In a similar way, immunohistochemical approaches such as c-fos labeling can also be used (but at a defined time point only) to study responses to suprathreshold stimuli.
models is that subtle side effects of drugs can be next to impossible to gauge, and so the preclinical approaches will primarily inform on efficacy. Consideration is also needed on what is being measured in animals. A large majority of studies are behavioral, in which the response to a stimulus is defined by the time or force to withdrawal of the animal at a certain intensity of the stimulus. This will be close to the threshold for “pain.” Thus, a drug may have reasonable efficacy against a pain of this level but lose or even gain effectiveness at higher intensities, more related to the pains that give rise to clinical problems. This problem can be overcome by recording neuronal activity in vivo (although potentially complicated by general anaesthesia), in which responses of defined sensory neurons, for example in spinal cord, thalamus, or other areas with somatosensory inputs, can be quantified using stimuli that would be above the threshold. Effects on threshold and suprathreshold responses can therefore be revealed. In a similar way, immunohistochemical approaches such as c-fos labeling can also be used (but at a defined time point only) to study responses to suprathreshold stimuli.
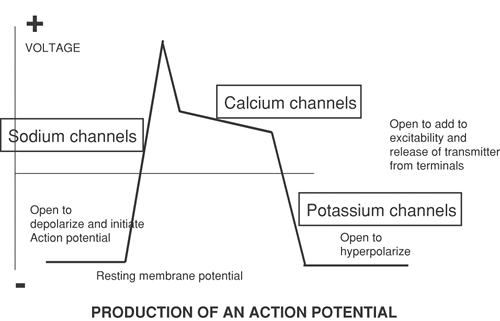 Figure 33-1. The contribution of ion channels to the action potential and the release of transmitter. |
Based on discoveries in animals and translation to humans, this chapter gives an account of ion channels in peripheral nerves related to pain as a basis for current and future potentially novel interventions based on the prevention of painful messages from entering the CNS.
Voltage-Gated Sodium Channels
Primary sensory neurons transmit nociceptive information from the periphery to the spinal cord. Following nerve injury, alterations in the excitability of neurons suggest changes in the expression and density of Na+ channels within the cell body and neuronal dendrites (9,10,11,12). Voltage-gated Na+ channels are responsible for the inward membrane current in the nervous system, required for the production of the continuously evoked action potentials vital for neuronal transmission of nociceptive and other sensory information (10). They are not only expressed abundantly on primary sensory neurons, but can also be separated into a large family of distinct Na+ channel subtypes encoded by a large variety of different genes within the DRG. They are also found to actively alter their expression throughout both development and disease. Some Na+ channels are specifically confined to sensory neuron populations, as opposed to other neural systems (13).
Voltage-gated Na+ channels are heteromeric protein complexes, composed of large α- (260 kDa) and smaller β-subunits (33–45 kDa). Each protein complex contains one α-subunit, forming the ion channel pore, and two β-subunits, named β1, -2, and -3 (12). Originally, there were thought to be eight separate VGSCs within the nervous system, all of which were encoded by eight different genes (9). However, more recent accounts specify nine α-subunit isoforms, named Nav1.1 through 1.9, using modern nomenclature (12) (see Table 33-1). Within the peripheral nervous system, a number of VGSCs have been found, including Nav1.6, 1.7, 1.8, and 1.9. These VGSCs are located on different nerve fiber types and as such, their distribution may alter following neuronal insult (9,12). As interest, both industrially and academically, has risen exponentially over the past few years toward the development of VGSC blockers as neuropathic pain drugs, so has nomenclature changed for a better understanding and a more uniform recognition of the large number of detected VGSCs now cloned (see Table 33-1).
Previously, in situ hybridizations and reverse transcription polymerase chain reaction (PCR) techniques, which reveal the messenger RNAs encoding distinct Na+ channels, revealed that within the DRG there were only six different Na+ channel transcripts expressed, namely the Nav1.1 through Nav1.6 channels. These Na+ channels were found in large quantities, mostly in medium and large DRG cell types. However these Na+ channels, which elicit Na+ channel currents, are all found to be expressed at varying levels in other neuronal cell types (9,10,12). The DRG also expresses Na+ channels that are exclusively or selectively expressed in the primary sensory neurons, as opposed to other neuronal populations; the Nav1.7, Nav1.8, Nav1.9, and NavX channels (9,12,13).
All of these Na+ channels can be divided based on their pharmacologic sensitivity to the toxin, tetrodotoxin (TTX), a noncompetitive voltage-gated Na+ channel blocker, into TTX–sensitive and TTX-resistant VGSC categories (9,12).
The TTX–sensitive Na+ channels include Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.7, whereas the rest are TTX-resistant Na+ channel types. Nav1.7 Na+ channel transcripts are also located within the terminals of the majority of DRG neurons (12). The Nav1.8 Na+ channel, as well as the Nav1.9 channel, is found in trigeminal neurons as well as small-diameter C-type DRG cells. It has emerged that Nav1.9 is also present in human embryonic kidney (HEK) cells as well. Nav1.8 and Nav1.9, which are TTX-resistant, have caused much interest, as the majority of nociceptive neurons make up small DRG neurons (9,10,11,12,13,14,15,16). Furthermore, it is known from more recent studies that Nav1.8 activates a TTX-resistant current within C-fiber populations, exhibiting slow-activating and inactivating, as well as rapid repriming kinetics (9,12) that would be suggestive of a role in generating abnormal patterns of firing. It is therefore evident that VGSCs are prevalent and participate in the conduction of nociceptive information. Not only is the development of action potentials in small DRG neurons largely diminished in Nav1.8-null mutant mice, but hypoalgesia also develops in response to application of varying forms of noxious stimuli (9,17). Nav1.9 is thought to be active at the resting membrane potential in small DRG cells, and it has therefore been suggested that Nav1.9 regulates the resting membrane potential of nociceptive C-fibers. Recent findings have located Nav1.9 in the hippocampus and therefore lay doubt to the initial hypothesis that Nav1.9 is restricted to the DRG (16).
The TTX–sensitive Na+ channels include Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.7, whereas the rest are TTX-resistant Na+ channel types. Nav1.7 Na+ channel transcripts are also located within the terminals of the majority of DRG neurons (12). The Nav1.8 Na+ channel, as well as the Nav1.9 channel, is found in trigeminal neurons as well as small-diameter C-type DRG cells. It has emerged that Nav1.9 is also present in human embryonic kidney (HEK) cells as well. Nav1.8 and Nav1.9, which are TTX-resistant, have caused much interest, as the majority of nociceptive neurons make up small DRG neurons (9,10,11,12,13,14,15,16). Furthermore, it is known from more recent studies that Nav1.8 activates a TTX-resistant current within C-fiber populations, exhibiting slow-activating and inactivating, as well as rapid repriming kinetics (9,12) that would be suggestive of a role in generating abnormal patterns of firing. It is therefore evident that VGSCs are prevalent and participate in the conduction of nociceptive information. Not only is the development of action potentials in small DRG neurons largely diminished in Nav1.8-null mutant mice, but hypoalgesia also develops in response to application of varying forms of noxious stimuli (9,17). Nav1.9 is thought to be active at the resting membrane potential in small DRG cells, and it has therefore been suggested that Nav1.9 regulates the resting membrane potential of nociceptive C-fibers. Recent findings have located Nav1.9 in the hippocampus and therefore lay doubt to the initial hypothesis that Nav1.9 is restricted to the DRG (16).
Table 33-1 Location of various sodium channel proteins in vertebrates | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Much interest has evolved around the concept of Na+ ion channels and their targets as prospective analgesics following nerve injury, because it is now apparent that the type of channels expressed following neuronal insult is altered (9). Recently, development of antisense oligonucleotides that disturb Nav1.8 synthesis have been used to “knock down” Nav1.8 VGSCs, resulting in a 50% reduction in the expression of these VGSCs (12,17). Interestingly, following spinal nerve ligation, “knock-down” has been seen to reduce neuropathic pain and its associated reductions in thermal and mechanical sensory thresholds. The same treatment in normal animals had no such effect on the sensory threshold values to peripherally applied noxious thermal stimuli (12,17). These knock-down studies have not found any effect of downregulation of the channel on thermal or mechanical hypersensitivity in neuropathic animals following attenuation of Nav1.9 synthesis. This is consistent with findings in sham operated animal groups (18).
Following neuronal axotomy, other studies have revealed that both an upregulation of the α-III Na+ channel and a downregulation of Nav1.8 and Nav1.9 gene expression occur within DRG neurons (9,19). Downregulation of Nav1.8 channel proteins in more recent studies following spinal nerve ligation of L5–L6 have specified that Nav1.8 is reduced in the injured DRG cell bodies; thus, Nav1.8 channel functioning is unlikely to occur in damaged nerve fibers (17,20,21). Such downregulation possibly prevents electrical conductivity in damaged C-fiber populations (12,17,22). This is despite original theories that, based on the role of Nav1.8 channels in DRG cell conductance, following nerve injury alterations in the activity of Nav1.8 protein channels could be responsible for spontaneous activity and repetitive firing of DRG cells (6,9). Interestingly, not only does such downregulation result in a reduction of TTX-resistant Na+ currents within the DRG neurons, but also in the development of a rapidly conducting repriming current within the TTX-sensitive Na+ currents in DRG neurons (6,9). This is thought to be due to the upregulation of the previously silent Nav1.3 Na+ channel. This latter channel relates to neuropathic pain and phantom phenomena that are seen after spinal cord injury (SCI), in which any molecular cause based on peripheral fiber ion channel changes cannot be likely due to the central location of the injury. Immunohistochemical investigations demonstrate abnormal expression of the Nav1.3 channel within central spinal cord dorsal horn neurons and also in the projection area of thalamic neurons after SCI. The changed expression of the channel may be a basis for abnormal hyperexcitability whereby denervated neurons start to act as amplifiers and generators (23,24).
Using the spinal nerve ligation (SNL) model of neuropathy, studies have also shown that the levels of Nav1.8 are unaltered in the uninjured L4 ganglia, yet analysis of the sciatic nerve reveals the levels of Nav1.8 are dramatically increased (21). In the neuropathic group, TTX was unable to block the compound action potentials (CAP) at C-fiber latencies, compared to sham operated rats whose CAP remained sensitive (>90%) to TTX (100 μm). These studies strongly
emphasize the possible role of Nav1.8 blocking agents in the reversal of neuropathic pain conditions (21). Following Nav1.8 knock-down studies in the sciatic nerve, it has become evident that Nav1.9 channels may be responsible for the observed residual TTXr currents in the C-fiber population (21). However, it is clear that the presence of Nav1.9 channels along the nerve axon is undisturbed by nerve damage, as differences in the density of voltage-gated Na+ channel currents in neuropathic and sham operated animals is minimal. Nav1.8 knock-down in uninjured neurons have also proven to be both antiallodynic and antihyperalgesic in relevant animal models (21). It is feasible that such alterations in Na+ channel expression, as well as changes in Na+ channel densities within DRG neurons, may underlie the spontaneous firing and lower thresholds that are responsible for such abnormal firing after axotomy and nerve damage (25,26). It is also possible that the development of spontaneous activity is a prerequisite of the hypersensitive allodynic and hyperalgesic states that characterize neuropathic conditions, along with the actual insult to the peripheral nerves (4,9,12).
emphasize the possible role of Nav1.8 blocking agents in the reversal of neuropathic pain conditions (21). Following Nav1.8 knock-down studies in the sciatic nerve, it has become evident that Nav1.9 channels may be responsible for the observed residual TTXr currents in the C-fiber population (21). However, it is clear that the presence of Nav1.9 channels along the nerve axon is undisturbed by nerve damage, as differences in the density of voltage-gated Na+ channel currents in neuropathic and sham operated animals is minimal. Nav1.8 knock-down in uninjured neurons have also proven to be both antiallodynic and antihyperalgesic in relevant animal models (21). It is feasible that such alterations in Na+ channel expression, as well as changes in Na+ channel densities within DRG neurons, may underlie the spontaneous firing and lower thresholds that are responsible for such abnormal firing after axotomy and nerve damage (25,26). It is also possible that the development of spontaneous activity is a prerequisite of the hypersensitive allodynic and hyperalgesic states that characterize neuropathic conditions, along with the actual insult to the peripheral nerves (4,9,12).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


