FIGURE 148.1 Schematic representation of the interrelationship of the absorption, distribution, binding, metabolism, and excretion of a drug and its concentration at its locus of action. Possible distribution and binding of metabolites are not depicted. (Adapted from Benet LZ, Kroetz DL, Sheiner LB. Pharmacokinetics: the dynamics of drug absorption, distribution and elimination. In: Hardin JG, Limbird LE, Gilman AG, et al., eds. Goodman and Gilman’s The Pharmacologic Basis of Therapeutics. 9th ed. New York: McGraw-Hill; 1996:3.)
In first-order kinetics, a plot of the logarithm of serum concentration after initial distribution versus time is a straight line. As seen in Figure 148.2, the IV injection of a drug results in an initially high serum drug concentration, followed by a rapid decrease because of drug distribution. After the distribution (or alpha) phase, the serum concentration further decreases because of the drug elimination (or beta) phase. At any point in time, the serum drug concentration (Cs) can be calculated from the biexponential disappearance function:

where α and β are the first-order rate constants for the alpha and beta phases, respectively (5).
Pharmacokinetic parameters of particular value in the application of pharmacokinetic principles to clinical practice include the volume of distribution, clearance, half-life, and bioavailability.

FIGURE 148.2 Schematic graph of serum drug concentrations (Cs) plotted on a logarithmic scale versus time after a single intravenous bolus injection. (Adapted from Greenblatt DJ, Koch-Weser J. Clinical pharmacokinetics. N Engl J Med. 1975;293:703.)
Volume of distribution (Vd) is the apparent volume of fluid in which a given dose would have to be distributed to achieve the observed serum concentration as mathematically described by the following equations:
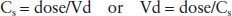
Clinically, the volume of distribution is useful for estimating the initial loading dose required to achieve a desired serum concentration or for calculating an incremental bolus dose required to raise a serum level by a desired amount. For instance, theophylline has a volume of distribution of approximately 0.5 L/kg in most patients; thus a 60-kg person would have an apparent volume of distribution of 30 L. If the desired serum concentration is 15 mg/L, the loading dose required to achieve this goal may be calculated as follows:

Clearance (Cl) is defined as the volume of blood or serum from which all drug is removed per unit of time. Clearance can be affected by alterations in distribution, metabolism, or excretion. Specific issues relating to changes in clearance from impairment of renal or hepatic function are discussed later in this chapter. At steady state, the drug administration rate equals the drug elimination rate (6). Based on this principle, drug clearance can be useful in determining the amount of drug required to maintain a therapeutic drug level. With an intravenously administered or completely absorbed oral agent, the steady-state serum concentration (Css) can be determined by the following equations:

Half-life (t1/2) is a function of both drug volume of distribution and clearance:

In most cases, this variable refers to elimination half-life, meaning the time required to reduce the initial serum concentration by 50% after the initial distribution phase. The drug half-life can be useful in determining the amount of time required to reach steady-state serum concentration. Four elimination half-lives are required to achieve approximately 94% of steady state. The time required to reach steady-state serum concentration is independent of dose and dosage interval. As seen in Figure 148.3, drug accumulation occurs with repeated dosing until equilibrium or steady state is reached at approximately four to five half-lives. If, in this example, the desired pharmacologic response is observed at a serum concentration of 1 to 2 units, a period of time after the initiation of therapy exists during which the serum concentration is subtherapeutic. To attain therapeutic levels at the initiation of therapy, a loading dose may be given, followed by maintenance doses.
For example, digoxin has an elimination half-life of 36 hours in a patient with normal renal function. Therefore, a period of 6 days is required to achieve a steady-state drug level with daily dosing. If a loading dose of 1 mg is incrementally administered over 24 hours, therapeutic digoxin concentrations may be achieved more rapidly.
The bioavailability of a drug is defined as the fraction of a drug dose that reaches the systemic circulation. Bioavailability is a consideration primarily with orally administered agents. Absorption can be affected by many factors relating to the pharmaceutical dosage form (e.g., tablet, capsule, oral liquid, suspension, or sustained-release product), gastric pH and emptying time, intestinal motility, and drug complex formation with other drugs or nutrients (7). Many orally absorbed drugs demonstrate a first-pass effect with significant metabolism in the liver before the drug enters the systemic circulation. Examples are propranolol, hydralazine, verapamil, and lidocaine. Lidocaine’s first-pass effect is so great that it must be administered by the parenteral route. For other drugs, such as propranolol and verapamil, oral doses need to be substantially higher than parenteral doses to account for this effect. For these drugs, the fraction of drug available to the systemic circulation or bioavailability (F) must be included in the calculation of steady-state serum concentration as follows:

Pharmacodynamics
Whereas pharmacokinetics is a detailed, often mathematical, approach to the way that the body handles a drug, pharmacodynamics is the discipline that defines the mechanisms of action of medications and their biochemical and physiologic effects on the patient (8). Although frequently discussed separately, in reality, pharmacokinetics and pharmacodynamics are closely interconnected. For example, along with the dosing regimen, the pharmacokinetic properties of a drug determine the concentration of that drug in the body, and in many situations, the drug concentration is directly related to the effects experienced by the patient. In addition, the degree of protein binding that a particular drug exhibits is typically considered a pharmacokinetic property. However, in many cases, it is only the unbound drug that is pharmacologically active and therefore responsible for the pharmacodynamic effects.
Drugs produce desired effects through various types of mechanisms of action. Some drugs demonstrate a relatively direct mechanism of action, such as mannitol administered for osmotic diuresis. When mannitol is administered intravenously, the osmolarity of the blood and other body fluids is increased, causing shifts in the distribution of water. Other medications, such as epinephrine and β-blockers, act by agonizing or antagonizing physiologic receptors. The effect of the drug is, therefore, directly related to the physiologic action of the associated receptor. Continued stimulation of some receptors with agonists frequently results in a state of downregulation. In the intensive care unit (ICU), where exogenous catecholamines are commonly used, receptor downregulation is important given that the continued or subsequent exposure to the same concentration of a particular agonist may have a lesser clinical effect over time. Other drugs, such as steroids, have very complex mechanisms of action involving crossing the cell membrane, regulating the transcription of specific genes, and ultimately altering protein synthesis. A practitioner’s knowledge of the mechanisms of action and physiologic effects of medications will help in determining appropriate therapy, anticipating pharmacodynamic drug interactions, and monitoring for therapeutic success.
SPECIAL POPULATIONS
Renal Impairment
Whether a pre-existing disease state or a new condition developed in the ICU, renal dysfunction is often present in critically ill patients. Reduced renal function may be related to a disease state, either iatrogenic or age related. Acute and chronic renal impairment can have profound effects on the pharmacokinetics and pharmacodynamics of many commonly used agents. In addition, the degree of renal dysfunction and resulting degree of fluid retention and uremia determine the degree of pharmacokinetic and pharmacodynamic changes. For example, a drug may exhibit no clinically significant changes in a patient with mild renal dysfunction, or the medication may be contraindicated in a patient with renal failure. The practitioner must evaluate each drug individually in relation to the degree of renal dysfunction present.

FIGURE 148.3 Fundamental pharmacokinetic relationships for repeated administration of drugs. (Adapted from Benet LZ, Kroetz DL, Sheiner LB. Pharmacokinetics: the dynamics of drug absorption, distribution and elimination. In: Hardin JG, Limbird LE, Gilman AG, et al., eds. Goodman and Gilman’s The Pharmacologic Basis of Therapeutics. 9th ed. New York: McGraw-Hill; 1996:23.)
Renal dysfunction has been shown to alter bioavailability, protein binding, volume of distribution, and excretion of certain compounds (9). There are relatively few data providing details regarding the alteration of bioavailability of most drugs in patients with renal dysfunction. However, certain conditions that are more common among uremic patients, including a higher gastric pH, altered gastric emptying time, vomiting, and intestinal edema, are known to alter the absorption and bioavailability of enterally administered drugs.
The distribution of some drugs may also be significantly altered in renal dysfunction. Because patients with significant renal dysfunction have a higher percentage of their body mass as water, the apparent volume of distribution for water-soluble drugs is often increased. In addition, the protein binding of drugs is often altered in the presence of uremia, which changes the apparent volume of distribution. In general, the plasma protein binding of acidic drugs, like phenytoin, is decreased (10). Because there is a larger percentage of unbound (free) drug available for pharmacologic activity, uremic patients may experience toxicity with total drug concentrations in the therapeutic range. For this reason, some practitioners prefer to measure free phenytoin, instead of total phenytoin, concentrations in patients with renal failure. Altered tissue binding also may affect the apparent volume of distribution of a drug. For example, digoxin’s volume of distribution has been reported to be significantly reduced in patients with renal disease (11). As a result, in patients with renal dysfunction, digoxin loading doses should be used cautiously and serum concentrations should be monitored carefully.
The major pharmacokinetic effect seen in renal dysfunction is the impaired elimination of drugs and their metabolites by the kidney. Renal clearance of pharmacologic agents is complex, involving one or more of the processes of filtration, tubular secretion, and reabsorption. Few drugs are excreted solely by glomerular filtration. Additionally, tubular secretion and reabsorption for many compounds may vary with the type of renal dysfunction and the administration of other agents (e.g., probenecid).
Clinicians must also consider that renal elimination in critically ill patients is a very dynamic process. Renal function can deteriorate very quickly when patients are hemodynamically compromised. Additionally, many agents frequently used in critically ill patients may cause renal dysfunction; radiographic contrast media, aminoglycoside antibiotics, amphotericin B, nonsteroidal anti-inflammatory drugs (NSAIDs), and calcineurin inhibitors are common examples (12). Changes in renal function may be acute or progressive and may or may not be reversible. Drug clearance requires constant reassessment in the ICU setting. It is essential to modify medication dosages in instances of renal compromise, and it is equally critical to adjust dosages as patients experience renal recovery to avoid therapeutic failure from suboptimal doses.
In evaluating medication use in renal impairment, the first step is to estimate the degree of dysfunction. Most medications that require dosage adjustment in patients with renal dysfunction are adjusted based on creatinine clearance. Although many hospital laboratories now report estimated glomerular filtration rate (eGFR) along with serum creatinine values, the National Kidney Disease Education Program recommends the use of equations such as the Modification of Diet in Renal Disease (MDRD) Study Equation, the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) Equation, and the Cockrtoft-Gault Equation to estimate glomerular filtration rate and creatinine clearance to guide drug dosing (13).
When evaluating a medication profile, the intensivist must evaluate each drug individually and determine if a dosage adjustment or additional monitoring is necessary. Although, for many medications, the initial or loading dose does not need to be altered in renal dysfunction, maintenance dosing regimens may need to be adjusted by prolonging dosing intervals, reducing the standard maintenance dose, or both, depending on the individual medication and degree of renal dysfunction. In cases of drugs with a narrow therapeutic index, serum concentrations should be monitored carefully to avoid potential toxicity. In general, steady state should be achieved before a serum concentration is obtained. If a therapeutic effect has not been achieved—as with uncontrolled seizures—or toxicity is suspected, an earlier sample may be warranted to assess the appropriateness of redosing or holding therapy. For example, in patients with normal renal function, a vancomycin trough level is typically obtained before the third or fourth dose; however, in the setting of renal dysfunction, a trough level prior to the second dose may be beneficial to help determine the patient’s ability to clear the drug before further dosing.
In addition to the parent drug, practitioners must also consider the elimination route of any metabolites. For example, the active metabolite of morphine, morphine-6-glucuronide, is renally excreted and may exert important clinical effects when it accumulates in patients with renal failure (14). Although this does not prohibit the use of morphine in patients with renal dysfunction, practitioners may want to consider another agent, such as fentanyl, that does not have an active metabolite (15). If morphine is the preferred drug, a longer duration of action should be expected in a patient with renal impairment when compared to the same dose in a patient with normal renal function. Frequent assessment of morphine use by pain score and sedation level should be planned.
Critical care practitioners must also be mindful of the effect of renal replacement therapy on drug clearance. The degree of drug clearance is determined by both drug-specific factors and the type of renal replacement therapy used. Drug-specific factors include molecular weight, solubility, degree of ionization, protein binding, and volume of distribution (9). For example, standard modes of dialysis are relatively ineffective in significantly clearing phenytoin, a highly protein-bound drug, from the body. However, more complicated techniques such as high-flux dialysis with charcoal hemoperfusion have been reported to remove considerable amounts of drug in a patient with phenytoin toxicity (16). Dialysis-specific factors that can affect the degree of drug clearance include the mode of dialysis, type of filter used, filter pore size, ultrafiltration rate, and blood and dialysate flow rates (17). As a general rule, peritoneal dialysis is less efficient in drug removal compared to intermittent hemodialysis and continuous renal replacement therapies (such as continuous venovenous hemodiafiltration) (9). Because of the potential complexity of drug dosing in the presence of renal replacement therapy, consulting a clinician or clinical pharmacist who is readily familiar with the pharmacokinetic implications of intermittent or continuous renal replacement is advisable.
Hepatic Disease
Like renal insufficiency, hepatic disease can also significantly alter the pharmacokinetic profile of a drug and the ultimate effect on the patient. Unfortunately, data relating the degree of hepatic dysfunction in relation to drug metabolism and elimination is limited, and dosage adjustment guidelines for hepatic dysfunction are available for a limited number of medications.
As patients with severe liver failure often suffer from secondary gastrointestinal problems, the bioavailability of some orally administered drugs may be altered (18). Severe liver failure and cirrhosis have been associated with delayed gastric emptying and delayed absorption (18). For example, enteral furosemide has been shown to have delayed absorption in patients with mild and severe cirrhosis irrespective of the presence of ascites (19). In addition, patients with cirrhosis are at increased risk for severe upper gastrointestinal tract bleeding, which can limit enteral medication tolerance (20).
Hepatic failure also affects the volume of distribution of some drugs. Patients with significant ascites comprise a larger portion of their body weight as water. Therefore, the volume of distribution of hydrophilic drugs is increased (18). Most patients with significant liver dysfunction are also hypoalbuminemic, leading to a higher free fraction of highly protein-bound drugs.
Many factors influence hepatic drug clearance; however, the most significant characteristics are hepatic blood flow and enzyme activity. High-extraction drugs are significantly removed (>60%) during the first passage across the liver, and the clearance of these drugs is significantly dependent on hepatic blood flow (18). In significant liver disease, such as cirrhosis, hepatic blood flow is decreased or totally shunted if portosystemic shunt is present (21). When a drug is administered enterally in these patients, drug enters the systemic circulation with minimal or no first-pass effect (22). Thus, the bioavailability of some orally administered drugs with extensive first-pass effect is greatly increased, and dosages should be appropriately reduced to prevent adverse side effects (23). Table 148.1 lists some of the high-extraction drugs that may be used in the ICU.
Low-extraction drugs are those that experience a relatively minimal first-pass effect (<30%), and initial bioavailability is not largely affected by liver disease (18). The clearance of hepatically metabolized, low-extraction drugs may be influenced by the effects of liver disease on the enzyme system responsible for the metabolic process. A reduction in the activity of drug-metabolizing enzymes in livers from cirrhotic patients is associated with increasing disease severity (24,25). However, a large variation in the degree of enzyme activity impairment exists, probably due to the heterogeneity of the enzymes involved in drug metabolism processes. Numerous enzymes are affected differently by liver disease. For example, oxidation reactions tend to be more sensitive than glucuronidation (26).
| TABLE 148.1 High-Extraction Drugs Commonly Used in the Intensive Care Unit |
 |
Low-extraction ratio drugs are less dependent on hepatic blood flow but are highly dependent on protein binding and the level of enzymatic capacity of the liver. These drugs are susceptible to changing clearance because of the induction or inhibition of liver enzymes and are sometimes referred to as enzyme limited or capacity limited (27). The hepatic clearance of some enzyme-limited drugs is termed binding sensitive or binding restrictive when only the circulating free drug is removed. Therefore, a decrease in drug binding causes an increase in the hepatic clearance of the drug. Drugs that are highly protein bound, such as phenytoin and warfarin, are most sensitive to this change in clearance.
Multiple factors determine the degree of impairment for the hepatic clearance of medications, and no comprehensive standard guidelines are available to aid practitioners in the dosage adjustments of these medications. In addition, most available data that addresses the effect of liver disease on drug clearance are from studies evaluating patients with a single-organ dysfunction (e.g., alcoholic cirrhosis); this is frequently not the case in critically ill patients. Therefore, any degree of other organ dysfunction, such as renal impairment or low cardiac output state, could further alter the pharmacokinetics and pharmacodynamics of the drug. The critical care team must initially evaluate the extent of organ dysfunction and determine if a dosage adjustment is likely to be needed, and then monitor the patient carefully for early signs of toxicity. When possible, blood concentrations of the affected medications should be monitored and monitoring of the free drug concentrations may be preferred. Dosing recommendations may be available for some medications based on the Child–Turcotte–Pugh (CTP) score. The labeling guidelines of selected medications offer dosing guidelines based on the CTP score and an FDA guidance statement included its use for pharmacokinetic studies in hepatic impairment (28). Clinicians should note that these recommendations are often based on chronic, rather than acute, liver failure. For medications without CTP-score-based recommendations, doses may be modified based on the basis of the drug’s protein binding and hepatic extraction ratio.
Pregnancy
Pregnant women represent a subset of critically ill patients with unique needs and characteristics. Clinicians must consider maternal physiologic changes and assess the risks and benefits for both mother and fetus as they formulate treatment plans. Each subspecialty discipline must share their expertise with the intensivist team, and when possible, decisions should be made collaboratively with both the mother and fetus in mind. Even in the otherwise healthy pregnant female, determining the optimal dosing regimen presents a considerable challenge due to the significant pharmacokinetic changes that occur during pregnancy.
Few gastrointestinal tract changes may affect drug absorption during pregnancy (29). With the exception of the active labor period, gastric emptying time appears to be unchanged. However, due to a decrease in gastrin secretion, pregnant women have a higher gastric pH and slower intestinal motility (30); the clinical significance of these changes in relation to drug absorption is not known. Some women experience significant nausea and vomiting, especially during the first trimester, which may affect their ability to tolerate some enteral medications. Noxious stimuli of any kind should be minimized.
Unlike absorption, the volume of distribution of certain drugs can be significantly changed by pregnancy. Most impressively, blood volume increases by approximately 40% to 50% during pregnancy (31). The exception is patients with severe pre-eclampsia or eclampsia; blood volume may expand very little in this population (29). Changes in plasma protein concentrations, such as a decrease in albumin concentration, can affect the free fraction of some drugs. Some examples of drugs that have a higher free fraction during pregnancy include diazepam, valproic acid, and phenytoin (32).
Pregnancy can also affect the metabolism and elimination of drugs as well. Interestingly, hepatic metabolism via the cytochrome P450 (CYP) system may be increased or decreased, depending on the specific enzyme. Pregnancy decreases the activity of CYP1A2 and CYP2C19. However, the activities of CYP3A4, CYP2D6, CYP2C9, and CYP2A6 are increased (30). The renal clearance of some medications is also significantly increased during pregnancy due to an increase in GFR of approximately 50% (30). For example, several β-lactam antibiotics, such as ampicillin, cefazolin, and piperacillin, have been shown to have increased clearance in pregnant women (33–35). Alterations in tubular secretion/reabsorption may also occur; however, few data are available in this area. Based on the available pharmacokinetic studies of renally excreted drugs, the effect of pregnancy on drug clearance varied widely from 20% to 65% above the clearance in nonpregnant women (30). For this reason, drugs that are primarily renally excreted unchanged may require a dosage increase of 20% to 65% to maintain prepregnancy blood concentrations (30).
In addition to pharmacokinetic changes that occur during pregnancy, the critical care practitioner must also consider other physiologic changes that may affect the patient’s need for drug therapy. For example, pregnant women are normally hypercoagulable and are at risk for venous thrombosis. The addition of other factors commonly seen in the ICU, such as immobility and endothelial injury, may put them at even higher risk. Unfractionated heparin and low–molecular-weight heparins (LMWHs) are the drugs of choice for the prevention or treatment of thrombosis during pregnancy. Practitioners should be aware that many pregnant women require higher doses of heparin compared to nonpregnant counterparts due to increased concentrations of heparin-binding proteins, increased volume of distribution, and increased clearance (36). In fact, the dose required for therapeutic anticoagulation may be as much as twice the typical weight-based dose (37). LMWHs are considered to be a safe alternative to unfractionated heparin during pregnancy, but their use in the acute setting for the treatment of venous thrombosis is controversial. Similar to unfractionated heparin, the clearance of LMWHs is increased during pregnancy, making dosing less straightforward than in the nonpregnant patient. Pregnant women have been shown to require higher than standard doses of LMWH to maintain goal antifactor Xa concentrations (37,38). Therefore, frequent monitoring of antifactor Xa concentrations is recommended, and multiple dosage adjustments may be required. With this increase in monitoring and related potential dosage adjustments, some argue that many of the advantages, both clinical and financial, of LMWHs are lost in this setting.
In summary, when the critical care practitioner is faced with determining the drug regimen for a pregnant woman, several steps should be taken. Most importantly, a multidisciplinary approach, including experts in the area of obstetrics and drug therapy, is invaluable. Next, the need to prescribe any drug must be carefully considered prior to the ordering process, and standard protocols should be modified as appropriate. Once the need for a drug is determined, the practitioner must consider the implications on fetal development and choose the safest drug available that will appropriately treat the mother. Historically, in considering the safety of a drug during pregnancy, many practitioners referenced the drug package labeling or an information database to view the pregnancy risk factor category (A, B, C, D, or X) used by the U.S. Food and Drug Administration to classify risk. In December of 2014, the FDA published the Content and Format of Labeling for Human Prescription Drug and Biological Products; Requirements for Pregnancy and Lactation Labeling, otherwise known as the “Pregnancy and Lactation Labeling Rule” or PLLR. This rule, effective June 30, 2015, mandated modifications to both the content and format for prescription drug labeling to assist clinicians in characterizing the relative safety of a medication during pregnancy and in counseling of pregnant and nursing mothers to empower them to make informed decisions. The pregnancy letter categories were removed, and the subsections of pregnancy, labor and delivery, and nursing mothers were replaced by pregnancy, lactation, and females and males of reproductive potential. The pregnancy section was expanded and includes a risk summary, clinical considerations, data and information for a pregnancy exposure registry. The lactation subsection offers information about drug transfer into breast milk and potential implications for the infant. The women and men of reproductive potential subsection include information about requirements for pregnancy testing, contraceptive recommendations and information about infertility. The PLLR mandates that the labeling for prescription medications be updated when information is outdated. Labeling for over-the-counter medications has not changed and is unaffected by the new rule.
In addition to utilizing the information provided in a medication’s package labeling, a thorough search for information should be conducted, and the current trimester of the pregnancy should be considered. Some medications are safer for use during certain periods of the pregnancy. For example, NSAIDs are thought to carry the most risk during the first and third trimesters (39). In addition to primary literature, a medical reference that specifically addresses medication use in pregnancy should be consulted (39). Once a treatment has been chosen, the practitioner must also consider the physiologic changes during pregnancy that may necessitate dosage adjustment and utilize therapeutic drug monitoring when available.
ADVERSE DRUG REACTIONS
The term ADRs encompasses a broad range of drug effects, ranging from exaggerated but predictable pharmacologic actions of the drug to toxic effects unrelated to intended pharmacologic effects (40). As such, the true incidence rate of ADRs is difficult to determine, with estimates ranging from 5% to 47% (41–43). There are patient specific as well as drug specific risk factors. Polypharmacy, the use of multiple medications or more medications than are clinically necessary, is likely the strongest risk factor. Clinicians must vigilantly monitor drug therapy regimens to minimize the number of medications, identify adverse outcomes early and discontinue offending agents as appropriate to optimize patient outcomes.
A small group of widely used drugs account for a disproportionate number of ADRs. Aspirin, anticoagulants, diuretics, digoxin, antimicrobials, steroids, and hypoglycemic agents account for 90% of reactions (40).
The most frequent ADRs result from the exaggerated, but predicted, pharmacologic actions of the drug and, as such, are readily identifiable and often preventable. Examples include hemorrhagic complications caused by anticoagulants, or hypoglycemia caused by oral hypoglycemic agents or insulin. The most important determinant for such adverse effects is an abnormally high drug concentration at the receptor site. This can occur for a variety of reasons, ranging from errors in administration, dose calculation, alterations in pharmacokinetics—such as reduction in the volume of distribution, rate of metabolism, or rate of excretion—or drug interactions resulting in increased concentration of free drug at the receptor site. Regardless of etiology, all have the potential to negatively impact the patient.
Other ADRs result from toxic effects unrelated to intended pharmacologic actions. Such events are, therefore, often unpredictable and frequently severe. Various mechanisms are involved: genetic, immunologic and nonimmunologic, or idiosyncratic. A list of genetic susceptibilities leading to ADRs with certain drugs is presented in Table 148.2. The most relevant examples are succinylcholine-induced prolonged apnea (suxamethonium sensitivity) and drug-induced malignant hyperthermia (MH).
Succinylcholine is a depolarizing neuromuscular blocking agent that is rapidly metabolized by plasma pseudocholinesterase, with a normal duration of blockade (paralysis) of 2 to 4 minutes. Approximately 1 in 3,200 patients is homozygous for a defective pseudocholinesterase, which has an autosomal recessive transmission and demonstrates markedly prolonged block with succinylcholine in the range of 3 to 8 hours (44).
MH is a clinical syndrome of muscle rigidity, tachycardia, tachypnea, rapidly increasing temperature, hypoxia, hypercapnia, hyperglycemia, hyperkalemia, hypercalcemia, lactic acidosis, and eventual cardiovascular collapse that occurs during, or after, general anesthesia (45). Several drugs have been implicated in triggering MH, particularly the halogenated inhalational agents (halothane, enflurane, and isoflurane), succinylcholine, and possibly an amino amide local anesthetic (lidocaine or bupivacaine) (46,47).
The cause of MH seems to be an inability of the sarcoplasmic reticulum of skeletal muscle to take up released myoplasmic calcium. When an MH episode is triggered, the myoplasmic levels of calcium rise tremendously, accelerating muscle metabolism and contraction, and leading to the clinical manifestations of the syndrome (45). The manifestation of MH may appear in the operating room or in the ICU shortly after the end of the surgical procedure.
Dantrolene is the cornerstone of therapy for MH (48). It reduces rigidity and restores muscle function by preventing calcium release from the sarcoplasmic reticulum and antagonizes its effects on muscle contraction (49). Treatment of MH is initiated by discontinuing the anesthetic and other possible triggering agents, ventilating the patient with 100% oxygen, providing hemodynamic support as needed, and administering dantrolene. The mortality rate from the fulminant syndrome was approximately 70% before the use of dantrolene (50). With early recognition and optimal therapy, reported mortality rates decreased from 10% to 7% (50).
An immunologically mediated ADR is epitomized by the classic anaphylactic reaction, which is IgE mediated, and a serum sickness–like condition. A similar reaction, but not immunologically mediated, known as anaphylactoid reaction, is sometimes seen as an indirect histamine release from the mast cells by morphine, or a complement activation leading to mediator release from the mast cell and basophil by aspirin and radiographic contrast media. There are a host of drug reactions for which the exact underlying mechanism of toxicity is not well understood, which are termed idiosyncratic reactions. Every organ system can potentially be adversely affected by drug exposures. Of the multisystem manifestations of ADRs, drug fever, drug withdrawal reactions, and anaphylaxis are worth elaborating.
| TABLE 148.2 Examples of Inherited Disorders Involving an Abnormal Response to Drugs |
 |
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


