Drug administration
Drugs are administered by many different routes. The aim of drug administration is to achieve therapeutic levels of the drug at its site (or sites) of action. In general, this is achieved using the vascular compartment as the transport mechanism for redistribution. Drug administration is, therefore, designed to produce suitable drug levels within the blood. The choice of route for a particular drug takes into account physical properties, target site of action, consideration of possible toxic effects and the practicalities of administration. The routes are summarised in Figure 27.2 in a practical classification. The enteral and topical routes are most easily accessible, but require absorption across a barrier or membrane to establish their effect.
| Enteral | Oral Buccal Rectal |
| Parenteral | Intravenous Intramuscular Subcutaneous Intradermal Transdermal Inhalational Transtracheal |
| Topical | Skin Eyes Ears Intranasal Vaginal Urethral |
Absorption
Absorption is the process of taking the drug from the site of administration to the blood. This is necessary for all enteral and parenteral routes except for intravenous (IV) administration. Systemic absorption may occur from topically administered drugs, but this is not the intended route. Absorption involves the crossing of barriers between administration site and vascular compartment with subsequent movement across the physical distance between the two. The distance within any local compartment is traversed by simple passive diffusion down the concentration gradient. Most barriers are made up of cells closely linked by tight junctions. The cell thus acts as both a filter and a device for active uptake, so the drug must pass into and then out of the cell to cross the barrier. The main principles affecting absorption include:
Simple passive diffusion
Facilitated diffusion
Active uptake
Pinocytosis
The rate of diffusion is shown by the following formula:
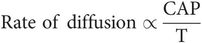
where
C = concentration difference either side of membrane
A = area of membrane
P = membrane permeability
T = membrane thickness
The ability of a drug (or any molecule) to pass through a membrane is influenced by:
Solubility in the membrane (lipid solubility)
Degree of ionisation
pH
Size of molecule
Carrier processes
Pinocytosis
Partition coefficient across the membrane
Enteral administration
Enteral administration encompasses any route that requires the gastrointestinal (GI) tract. Where a patient is incapacitated, access to the stomach and small intestine can still be achieved using a nasogastric tube or an endoscopically sited nasoduodenal small-bore feeding tube. Most enterally administered drugs have sites of action distant from the GI tract and therefore require absorption first. This exposes them to the first-pass effect, which is elimination during passage through the liver via the hepatic portal vein. The hepatic portal vein receives blood from the GI tract from oesophagus to upper rectum. Oral and nasogastric administrations imply that the drug enters the stomach, but absorption may occur throughout the length of the GI tract. Buccal administration, and to a degree rectal administration, bypass the liver and so avoid the first-pass effect. Enterally administered drugs may also be subjected to other effects before absorption, including neutralisation of alkaline drugs by gastric acid and enzymatic action in the intestinal lumen or wall. The advantages and disadvantages of enteral administration are as follows:
Advantages
Easy to give
Special equipment not required
Patient can self-administer
Disadvantages
May not be appropriate route
Patient may be unable to swallow
Drug may be destroyed in the GI tract lumen
May have excessively high first-pass effect
Drug may be irritant to GI tract
First-pass effect and bioavailability variable
Effective dose unpredictable
Most drugs administered enterally require absorption followed by distribution via the blood to the effector sites. It is difficult to measure effector levels of drug, but blood levels can be used as a predictor of achievement of an effective level. Absorption is therefore the mechanism for achieving an effective plasma level, and absorption and uptake are essential to this aim. For IV administration, however, this stage is bypassed. IV administration involves administration of a bolus of drug that rapidly becomes distributed throughout the whole circulating volume.
First-pass effect
Enterally administered drugs absorbed from the stomach, small intestine or colon enter the hepatic portal vein. The whole of the absorbed drug dose passes through the liver and is therefore potentially subjected to hepatic metabolism and extraction (elimination) before entering the systemic circulation. This is termed the first-pass effect.
Bioavailability
Bioavailability is the amount of drug administered by a given route that reaches the systemic circulation. IV administration therefore achieves 100% bioavailability. Bioavailability is usually taken to refer to oral administration (oral bioavailability), but other routes also have a bioavailability. Bioavailability is reduced by destruction in the lumen, poor absorption and metabolism. Oral bioavailability is greatly influenced by the first-pass effect, so that a high first-pass effect produces a low bioavailability.
Parenteral administration
By definition, parenteral administration uses routes other than the GI tract. Following absorption, the drug is transported in the blood. This may be in the plasma or the red blood cells or both, and may involve protein binding. The absorption process is generally simpler than enteral absorption but still entails some degree of variability between patients, sites and drugs. The advantages and disadvantages of parenteral administration are as follows:
Advantages
Unaffected by first-pass metabolism
Plasma levels may be more predictable
Does not require functioning GI tract
Does not require patient assistance
Disadvantages
Administration requires training
Not usually self-administered
Administration usually requires injection
Requires special equipment
Intramuscular
Intramuscular (IM) injection is one of the commonest methods of parenterally administering drugs, avoiding the need to cannulate a vein and slowing the onset of the effect, which may confer a safety advantage. The absorption profile means that the drug will be absorbed slowly over a period and will last longer than the same drug administered intravenously.
The factors affecting absorption from an IM injection are:
Drug solubility in blood
Tissue binding
Protein binding
Blood flow to site
Transdermal
Fentanyl and glyceryl trinitrate (GTN) are examples of drugs that may be administered transdermally. The method avoids the first-pass effect, and provides a slow absorption and therefore a prolonged effect without significant peaks and troughs in plasma levels. Transiderm-Nitro 5 has a reservoir of 25 mg GTN with a contact surface area of 10 cm2, which achieves an average 24-hour absorption of 5 mg. A plateau in the plasma level of glyceryl trinitrate is achieved within 2 hours of application. This plateau level is directly proportional to the surface area of the permeable membrane of the patch. It is important, therefore, that the patch makes good contact with the skin. Transiderm-Nitro 10 achieves double the transfer (10 mg per 24 hours) by doubling the contact surface area to 20 cm2.
Inhaled
Inhaled anaesthetic agents are covered elsewhere (Chapter 29). Their effect depends on achieving sufficient brain concentration of the drug (which is produced from an alveolar concentration) secondary to an inhaled dose at the nose or mouth.
Inhaled bronchodilators act in a topical manner, but systemic absorption also occurs.
Distribution
Once a drug is absorbed into the circulation it undergoes distribution. An overview of mass drug movement is shown in Figure 27.3.
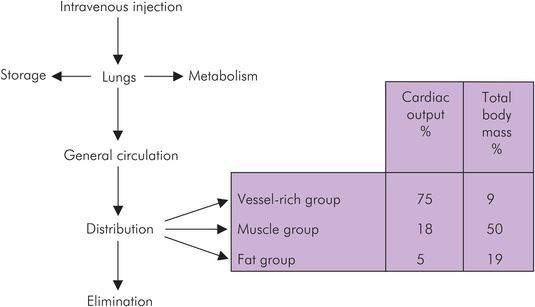
Overview of drug distribution
The distribution to individual tissues depends on the solubility of the drug in those tissues and its delivery to them. The factors determining the uptake of the drug and speed of distribution to an individual tissue are:
Plasma protein binding
Blood flow to tissue
Mass of tissue to which distributed
Tissue/blood partition coefficient
Tissue protein binding
Facilitated transport
Drug ionisation
Drug molecular size
Blood flow to tissues
Blood flow determines the speed of distribution and the amount of drug reaching the tissue capillaries, and therefore the amount available for uptake into that tissue.
Three compartments are generally used to explain pharmacokinetic principles: the vessel-rich group (VRG), the muscle group (MG) and the fat group (FG). The vascularity of these tissue groups varies, and the proportion of cardiac output received by them is shown in Figure 27.3.
Drug uptake by tissues
Uptake depends on the drug concentration in tissue and blood, and the partition coefficient. The blood concentration will change as it flows through the capillaries provided that tissue and blood are not at equilibrium. The drug will pass into (or out of) the tissue towards equilibrium. The commonest mode of transit is diffusion, but carrier-facilitated diffusion and active transport systems also occur. The partition coefficient indicates the ratio of drug concentration in neighbouring tissue compartments at equilibrium, and drug movement will occur towards this equilibrium point. As blood concentration falls due to redistribution and elimination, then the balance will be restored by movement out of the tissue back into the blood. The speed of transfer to and from compartments depends on the degree of deviation from the equilibrium partition coefficient and the ease of molecular movement. The mass of tissue involved will alter the rate at which equilibrium is reached and so in turn will alter the rate of uptake. For a given partition coefficient, a large volume of tissue will increase both total uptake and rate of uptake of the drug, but will reduce the speed of achievement of equilibrium.
The ease of transfer between compartments is influenced by:
Ionisation (determined by pH and pKa)
Membrane components
Molecular size
Figure 27.4 shows the temporal relationship of drug distribution within various compartments following an IV bolus administration of a typical IV induction agent.
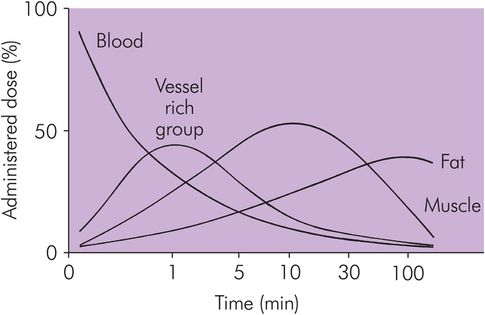
Drug distribution in various tissues against time following IV bolus administration of thiopental
Drug distribution causes the blood concentration to fall. The VRG then has more drug than plasma, so movement is reversed and the drug moves from the tissue back into the blood. At this early stage, the blood concentration is higher than that in either the muscle group or the fat group. There is a net redistribution from VRG to MG and FG. Later the same effect will occur from MG to FG, and much later the drug in FG will return to the blood and be eliminated. All redistribution processes require the blood as an intermediary phase.
Active transport
Penicillin is a drug that is actively transported. This is a unidirectional process regardless of the concentration gradient. Probenecid blocks active transport in the liver, kidney and choroid plexus, which increases the proportion in the blood, and so the apparent volume of distribution is reduced.
Protein binding
Protein binding alters the concentration of free drug as a proportion of the total. Different proteins have different affinities for drugs. Albumin mostly binds acidic drugs such as aspirin, whereas α1-glycoprotein is more important for basic drugs. The amount of drug bound is influenced by plasma pH (which alters the ionisation of the drug), age and certain disease states. Hypoalbuminaemia is a common finding in chronic disease, and in hepatic (reduced synthesis) and renal (increased loss) failure in particular. Such conditions reduce the amount of albumin available for drug binding, and therefore increase the amount of free drug. Note that α1-glycoprotein is increased in:
Obesity
Trauma
Burns
Postoperative period
Myocardial infarction
Carcinoma
Inflammatory diseases
Placental transfer
Most drugs cross the placenta to some degree. Transfer is favoured by high lipid solubility and a high un-ionised proportion. Placental blood flow and metabolism also influence transfer. The ratio of concentrations in fetal and maternal placental blood is called the feto-maternal concentration ratio (Figure 27.5).
| Induction agents | Propofol | 0.65–0.85 |
| Thiopental | 0.4–1.1 | |
| Volatile agents | Isoflurane | 0.7–0.91 |
| Nitrous oxide | 0.83 | |
| Opioids | Alfentanil | 0.3 |
| Fentanyl | 0.1–1 | |
| Morphine | 0.92 | |
| Pethidine | 1 | |
| Muscle relaxants | Suxamethonium | 0.04 |
| Atracurium | 0.05–0.2 | |
| Vecuronium | 0.11–0.12 | |
| Local anaesthetic agents | Bupivacaine | 0.2–0.4 |
| Lidocaine | 0.5–0.7 | |
| Ropivacaine | 0.2 |
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


