Pediatrics
16-1 Croup
Christy Salvaggio
Clinical Presentation
Croup typically affects children between the ages of 6 months and 6 years, with a peak incidence at 2 years of age. Croup occurs year-round but is most common in the fall and early winter. Male children are affected almost 1.5 times more often than female children.1 The onset of the illness is marked by prodromal symptoms typical of a viral upper respiratory tract infection (URTI), such as a runny nose, congestion, and mild cough. Fever is common. On approximately day 3 of the illness, the classic symptoms of croup develop, including a barky cough that may sound like a barking seal, hoarseness, and varying degrees of stridor with possible respiratory distress. The severity may vary greatly. The majority of patients have only a barky cough and URTI symptoms. Most of these patients never seek medical attention and are treated with supportive care at home. Patients with more severe disease have stridor when crying or agitated, and those most severely affected have stridor at rest, with marked increased work of breathing, hypoxia, or both. Classically, croup symptoms are exacerbated at night, and the second night of croup symptoms is often worse than the first night. Children typically improve if they are exposed to cool or humidified air while crying; other distress exacerbates the symptoms.
Pathophysiology
Croup, or acute laryngotracheobronchitis, is a viral infection most commonly caused by parainfluenza virus. It manifests with URTI symptoms and varying degrees of a barky cough, hoarseness, and stridor. Parainfluenza type 1 is the main cause of croup; however, other viruses are occasionally identified, including respiratory syncytial virus, parainfluenza types 2 and 3, influenza A and B, and adenovirus. Bacterial causes (e.g., Mycoplasma pneumoniae) are rare.1 Croup causes respiratory distress secondary to subglottic edema. Older children infected with parainfluenza or other croup-causing viruses are likely to present with laryngitis. Younger children manifest symptoms of croup because of the smaller diameter of their upper airway. Croup is rare before 6 months of age, probably because of the presence of maternal antibodies.
 FIGURE 16-1 A: “Steeple sign.” (Courtesy of Robert Hendrickson, MD.) B: Croup. The airway is smoothly narrowed or tapered (arrowheads). As a result, the hypopharynx is dilated (p). (From Ovassapian, with permission.) |
Diagnosis
Croup is a clinical diagnosis. Patients classically present with URTI symptoms, a barky cough, and stridor that are not difficult to diagnose. For patients with unusual presentations, those with severe symptoms not responding to treatment, or croup in a child older than 6 years of age, additional work-up may be required to rule out other upper airway obstruction. A complete medical history, including any history of intubations, recent choking episodes, or the presence of hemangiomas, should be elicited. The differential diagnosis for croup includes foreign body obstruction, airway hemangiomas, epiglottitis, tracheitis, and anterior mediastinal masses. A soft-tissue lateral neck radiograph to evaluate for possible upper airway obstruction may be helpful; however, in children with classic croup symptoms, a posteroanterior neck radiograph looking for the classic “steeple sign” is not necessary or recommended.
Acute spasmodic croup should also be considered as a possible diagnosis. It affects children 3 months to 3 years of age. Its presentation is almost identical to that of viral croup, with the acute onset of hoarseness, a barky cough, and inspiratory stridor; however, patients with spasmodic croup lack the prodrome of infectious URTI symptoms that includes fever.
Clinical Complications
Croup is usually a self-limited illness that resolves within 1 week. More than 90% of children with croup never develop symptoms more severe than mild stridor and dyspnea. In more severe cases, patients may have severe respiratory distress secondary to marked subglottic edema and inflammation; however, respiratory failure secondary to croup is rare. Viral laryngotracheobronchitis may predispose patients to secondary bacterial infections, including bacterial tracheitis.
Management
Mild cases of croup, involving a well-appearing child with URTI symptoms and a barky cough without stridor, may be treated with supportive care (e.g., cool or humidified air). Placing the child in the bathroom with a hot running shower may provide the humidified air. Alternatively, in cool weather, the child may be taken outside or placed near an open window to be exposed to cool air. Stridor that is present only with crying or other distress should be treated with a one-time dose of oral dexamethasone. Some studies have shown equal efficacy with lower Decadron doses (0.15 to 0.3 mg/kg). Oral Decadron is as efficacious as intramuscular Decadron; the latter should be reserved for patients who are unable to take oral medications.2 Nebulized budesonide, 2 to 4 mg, may be used instead of or in addition to Decadron.
Children who have stridor at rest are more concerning and need urgent care. Few data support the use of cool humidified air in the emergency department setting; however, it may be tried as long as it does not cause more distress in the child. Subcutaneous (s.c.) racemic epinephrine, 0.05 mL/kg to a maximum of 0.5 mL, should be administered immediately. Alternatively, 0.5 mL of 1:1000 of L-epinephrine may be administered, with equal efficacy.1 Oral or intramuscular Decadron should also be given. Racemic epinephrine may provide an acute improvement in symptoms, but the improvement is transient and patients’ symptoms may return or “rebound,” sometimes with increased intensity. All patients who require s.c. racemic epinephrine should be monitored for at least 3 hours.1 Well-appearing patients who have adequate follow-up and no stridor at rest may be discharged after 3 hours of observation. Any patient who has persistent stridor at rest or who requires a second dose of s.c. racemic epinephrine should be admitted for observation and s.c. racemic epinephrine on an as-needed basis. The s.c. racemic epinephrine may be administered as often as every 30 minutes for severe symptoms, but multiple doses should be administered with caution. Patients require careful monitoring for side effects.
REFERENCES
1. Wright RB, Pomerantz WJ, Luria JW. New approaches to respiratory infections in children: bronchiolitis and croup. Emerg Med Clin North Am 2002;20:93-114.
2. Rittichier KK, Ledwith CA. Outpatient treatment of moderate croup with dexamethasone: intramuscular versus oral dosing. Pediatrics 2000;106:1344-1348.
16-2 Bronchiolitis
Maria McColgan
Clinical Presentation
Bronchiolitis begins as an upper respiratory tract infection with rhinorrhea and mild cough, which may progress to respiratory compromise. Young infants may present with apnea. Patients may also present with feeding intolerance and signs of dehydration. Clinical signs include fever of 38.5 to 39° C (101 to 102° F), tachypnea, inspiratory and expiratory wheezing, crackles, retractions, hypoxemia, nasal flaring, and grunting. In severe cases, respiratory failure may ensue.
Pathophysiology
Bronchiolitis is an acute, inflammatory, obstructive disease of the bronchioles, often caused by a viral infection, in children younger than 2 years of age. Respiratory syncytial virus (RSV) is the leading cause of bronchiolitis (up to 80% of cases). Other causes include influenza, adenovirus, parainfluenza, and mycoplasma. Winter is the peak season for bronchiolitis. The peak incidence is between 3 and 6 months of age.1,2,3
 FIGURE 16-2 Note respiratory distress with substernal retractions in this child with bronchiolitis. (© David Effron, MD, 2004. Used with permission.) |
Diagnosis
The diagnosis is based on clinical findings. A chest radiograph should be obtained if clinically indicated, to rule out bacterial pneumonia. Typical radiographic findings include hyperinflation, hyperlucency, and flattened diaphragm. Patchy, interstitial infiltrates, representing areas of atelectasis, may be seen in 30% of patients. The white blood cell count usually is normal. Viral cultures usually are not helpful.1,2,3
Clinical Complications
Respiratory failure from hypoxemia or hypercarbia may occur. Mortality is highest in infants younger than 2 months of age, premature infants, and infants with bronchopulmonary dysplasia or congenital heart disease. Bronchiolitis may be complicated by secondary bacterial pneumonia or bacterial infection, especially in hospitalized infants.
Management
Treatment is supportive, including hydration, supplemental oxygen, and mechanical ventilation if necessary. Nebulized albuterol, racemic epinephrine, or both are commonly used. However, studies have shown only brief improvement in clinical score, and no effect on the duration of illness or hospitalization, with the use of these agents.1,2,3 Corticosteroids are generally considered not effective in bronchiolitis. Antibiotics should be used only if secondary bacterial infection is suspected. Criteria for hospital admission typically include respiratory rate greater than 60 breaths per minute, apnea, color changes, hypoxia, and inability to take adequate oral solutions. Hospitalization should be strongly considered for high-risk infants, including infants younger than 2 months of age, those with cardiac or pulmonary disease, and those who were born prematurely.1,2,3
REFERENCES
1. Flores G, Horowitz RI. Efficacy of beta2-agonists in bronchiolitis: a reappraisal and meta-analysis. Pediatrics 1997;100:233-239.
2. Dobson JV, Stephens-Groff SM, McMahon SR, et al. The use of albuterol in hospitalized infants with bronchiolitis. Pediatrics 1998; 101:361-368.
3. Garrison MM, Christakis DA, Harvey E, et al. Systemic corticosteroids in infant bronchiolitis: a meta-analysis. Pediatrics 2000;105:e44.
16-3 Diphtheria
Mark Silverberg
Clinical Presentation
Diphtheria classically manifests in children 1 to 9 years of age, but it may be seen in nonimmunized adults.1 The disease has an insidious onset, beginning with a sore throat and low-grade fever accompanied by nausea, vomiting, and diarrhea. The pharynx is initially red, but grayish-white lesions develop and coalesce into a single membrane covering the tonsils, soft palate, and uvula within 24 hours.1 This membrane is not easily removed and bleeds when forcefully removed.
Pathophysiology
Corynebacterium diphtheriae is a club-shaped, gram-positive rod with an incubation period of 2 to 5 days. Toxigenic strains produce diphtheria toxin that inactivates the translocase of ribosomes, preventing translation of messenger RNA into cellular peptides.1
 FIGURE 16-3 Pharyngeal pseudomembrane in diphtheria. (Copyright CW Leung, Princess Margaret Hospital, Lai Chi Kok, Kowloon, Hong Kong.) |
Diagnosis
The diagnosis of diphtheria is clinical, based on the presence of the classic gray/white pseudomembrane covering a mucosal surface. In every suspected case, samples should be cultured on Loeffler’s medium and analyzed for virulence by a gel diffusion test for toxigenicity.1 Diphtheria may develop on any mucosa surface, giving rise to nasal, laryngeal, and cutaneous diphtheria. Cervical lymphadenopathy usually is present. In cases of moderate disease, symptoms begin to resolve in 5 to 6 days even without the use of antitoxin, although it does increase the rate of symptom resolution.1 In severe disease (“bullneck diphtheria”), the symptoms are very severe and the entire neck becomes swollen, giving rise to its characteristic shape with airway compromise a danger.
Clinical Complications
Dehydration is often present due to dysphagia. Airway obstruction may develop and may progress to death in severe cases. Myocarditis is commonly encountered, and degenerative changes occur in the central nervous system in almost all fatal cases.1 Serum sickness may occur after treatment with the horse antitoxin.1
Management
Airway management is paramount in all suspected cases of pharyngeal diphtheria. Intravenous hydration should be initiated to prevent dehydration. Patients with suspected diphtheria should be placed in isolation and should receive diphtheria antitoxin.1 Penicillin and erythromycin are the drugs of choice; either is active against C. diphtheriae as well as streptococcal species, which are found in a large proportion of diphtheria cases.
REFERENCES
1. Hodes HL. Diphtheria. Pediatr Clin North Am 1979;26:445-459.
16-4 Pertussis
Rakesh D. Mistry
Clinical Presentation
Pertussis has three phases—catarrhal, paroxysmal, and convalescent. The catarrhal stage lasts approximately 1 to 2 weeks; symptoms resemble those of a simple upper respiratory tract illness. The paroxysmal stage is characterized by harsh, prolonged coughing spells, or paroxysms. It is during this stage that patients develop the classic “inspiratory whoop.” This inspiratory sound is usually limited to older toddlers and children. Infants and young children with pertussis cannot generate sufficient inspiratory strength to create this “whoop.” The paroxysmal stage typically lasts 1 to 2 weeks, after which symptoms wane. The convalescent stage of pertussis infection may last several weeks. Fever is absent in most cases of pertussis.1
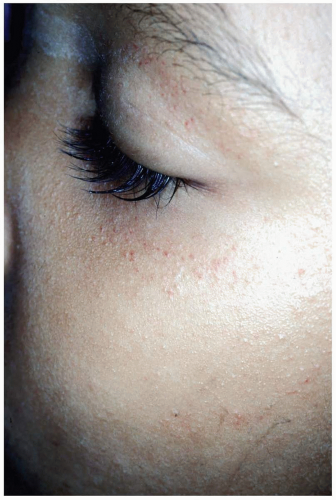 FIGURE 16-4 Facial petechiae are common in patients with pertussis as a result of continued, forceful coughing. (© David Effron, MD, 2004. Used with permission.) |
Pathophysiology
The organism responsible for pertussis is the gram-negative rod, Bordetella pertussis. B. pertussis has selective tropism for ciliated epithelial cells of the human respiratory tract, and local inflammation is the rule; invasive infection is not characteristic. Transmission is by aerosolized droplets, with transmission rates approaching 80% in close contacts.1
Diagnosis
Diagnosis is usually made during the paroxysmal stage. A complete blood count showing an absolute lymphocytosis supports the diagnosis, and the degree of lymphocytosis correlates with disease severity. The gold standard for diagnosis is isolation of B. pertussis in culture. This is accomplished by collecting mucus from the posterior nasopharynx with a Dacron or calcium alginate swab (cotton inhibits growth of B. pertussis). Direct fluorescent antibody (DFA) testing is the preferred modality for identification of B. pertussis in many institutions.
Clinical Complications
Management
Treatment of active pertussis infection in all stages consists of oral erythromycin. Erythromycin decreases infectivity of the host, but it diminishes disease severity only if it is instituted during the catarrhal stage. Patients younger than 12 months of age are at greatest risk for complications from pertussis infection, and inpatient management is prudent.1,2
REFERENCES
1. Waggoner-Fountain L, Hayden GF. Pertussis in primary care practice: recent advances in diagnosis, treatment, and prevention. Pediatr Clin North Am 1996;23:793-804.
2. Vitek CR, Pascual FB, Baughman AL, Murphy TV. Increase in deaths from pertussis among young infants in the United States in the 1990s. Pediatr Infect Dis J 2003;22:628-634.
16-5 Measles
Mark Silverberg
Clinical Presentation
Patients with measles may present with a prodrome of fever, cough, coryza, malaise, and conjunctivitis. Before 1968, an inactivated vaccine was used to prevent measles, and patients immunized before this date may present with “atypical measles,” which includes a magnified prodrome and a rash that spreads centripetally.1 An incubation period of 6 to 19 days exists between the time the viral infection occurs and the beginning of the classic clinical prodrome.1
Pathophysiology
Measles is caused by a highly contagious single-stranded RNA paramyxovirus and is spread via respiratory droplets. The virus multiplies in the respiratory mucosa and is then carried to the bloodstream in lymphoid cells.1 In 2001, 30 million cases of measles were reported worldwide; however, the disease is relatively uncommon in the United States since the introduction of a two-dose immunization program in 1963.1
Diagnosis
The diagnosis usually is made clinically. However, a number of serologic tests exist to confirm clinical suspicions.1 Koplik’s spots are pathognomonic for measles. Koplik’s spots (white spots that look like “grains of sand on a red background”) appear on the buccal mucosa approximately 2 days before the rash appears.1 The classic measles rash is maculopapular in nature; it starts on the face and progresses down the body. It then becomes brownish and fades in the same order as it appeared.
Clinical Complications
Complications of measles may occur in healthy persons but are more common in the very young, the malnourished, and those with suppressed immunity (e.g., patients with human immunodeficiency virus infection [HIV], those undergoing radiotherapy or chemotherapy).1 Complications include pneumonia, diarrhea, otitis media, premature labor, croup, hepatitis, encephalitis, seizures, and death.1,2 Other complications include idiopathic thrombocytopenia and subacute sclerosing panencephalitis, which usually is fatal and occurs 7 to 10 years after the initial measles infection.1
Management
The management of measles is supportive. Antipyretics should be given to prevent febrile seizures, and the patient should be encouraged to increase oral intake to prevent dehydration. Many patients become sensitive to light and benefit from muted lighting. Some authors believe that vitamin A helps decrease the complication rate associated with measles, but this is believed to be helpful only in individuals who are vitamin A deficient.1 Measles is a notifiable disease.1
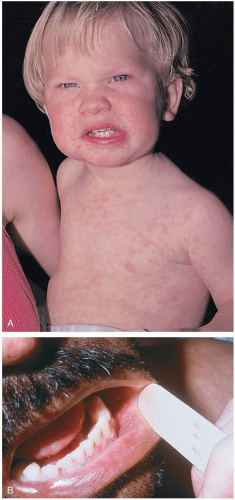 FIGURE 16-5 A: Skin rash of measles. Note periorbital edema and photophobia. (From Ostler, with permission.) B: Koplik’s spots in an adult patient. (From Goodheart, with permission.) |
REFERENCES
1. Stalkup JR. A review of measles virus. Dermatol Clin 2002;20:209-215.
2. Rall GF. Measles virus 1998-2002: progress and controversy. Annu Rev Microbiol 2003;57:343-37.
16-6 Mumps
Michael Greenberg
Clinical Presentation
Patients with mumps may present with prodromal symptoms including malaise, fever, chills, headache, and sore throat. This prodrome is followed by uncomfortable swelling of one or both parotid glands. Mumps orchitis, a common complication of the disease, manifests with unilateral pain and swelling of the scrotum. Approximately 30% of cases are subclinical and asymptomatic.1,2,3
Pathophysiology
Mumps is caused by the mumps paramyxovirus, which may invade tissues including the parotid glands, testicles, pancreas, and eyes.1,2 The period for incubation is 2 to 3 weeks, and the patient is contagious for approximately 10 days, from a few days before swelling is noted until the swelling resolves. Death from mumps is not a common event, but it is more common in young adults.1,2
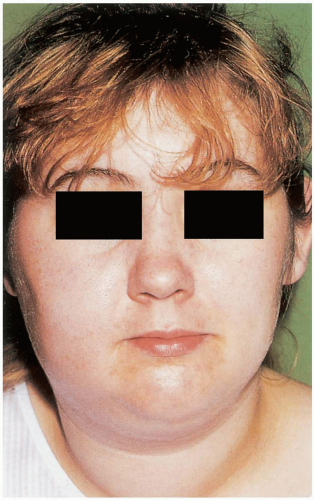 FIGURE 16-6 Parotitis (mumps). This teenager has bilateral parotid swellings as a result of mumps. (From Benjamin, with permission.) |
Mumps orchitis, which is common in preadolescent boys, is usually unilateral in nature. It rarely causes sterility unless the condition is bilateral. Mumps orchitis should be considered in the differential diagnosis for any patient who presents with scrotal swelling. Mumps is also one of the most frequent causes of unilateral hearing loss.1,2,3
Diagnosis
The diagnosis is based on clinical examination. Patients usually manifest fevers up to 103° F in conjunction with nonindurated swelling in the area of the parotid glands. The area overlying the parotids usually is not red or warm, unless a secondary bacterial parotitis has developed. In many cases of mumps parotidis, Stensen’s duct is erythematous and swollen. In some cases, facial edema spreads to include the anterior chest, resulting in obvious presternal edema. The diagnosis of mumps may be confirmed by serologic testing. Viral cultures may be obtained in some cases.1,2,3
Clinical Complications
Complications include orchitis, pancreatitis, unilateral hearing loss, cerebellar ataxia, cranial neuritis, meningitis, cerebritis, cerebellitis, polyradicular neuritis, encephalitis, transverse myelitis, spontaneous abortion, nephritis, and myocarditis. Among patients who develop the rare complication of meningoencephalitis, the mortality rate is approximately 2%.1,2,3
Management
Treatment is primarily supportive and directed at preventing dehydration and relieving discomfort. Scrotal pain may be addressed with scrotal support, cold compresses, and antiinflammatory medications. If dehydration has occurred, or if serious complications such as central nervous system infection are suspected, the patient should be promptly hospitalized.
REFERENCES
1. McQuone SJ. Acute viral and bacterial infections of the salivary glands. Otolaryngol Clin North Am 1999;32:793-811.
2. Nussinovitch M, Volovitz B, Varsano I. Complications of mumps requiring hospitalization in children. Eur J Pediatr 1995;154:732-734.
3. Kabakus N, Aydinoglu H, Yekeler H, Arslan IN. Fatal mumps nephritis and myocarditis. J Trop Pediatr 1999;45:358-360.
16-7 Rubella
Christy Salvaggio
Clinical Presentation
Children with rubella first present with a pruritic, maculopapular, erythematous rash that starts on the face and spreads to the extremities.1 The rash typically lasts 3 days, with the face clearing first. Adults may present with prodromal symptoms (fever, malaise, cough, sore throat, lymphadenopathy) several days before the rash appears. Lymphadenopathy lasts about 1 week and is most prominent in the posterior auricular, suboccipital, and posterior cervical chains. Arthralgias and arthritis, infrequently seen in children, are more common in adolescents and adults, especially women.
Pathophysiology
Humans are the only host for the RNA togavirus that causes rubella. Transmission is primarily by nasopharyngeal, airborne, or droplet spread. Patients are infectious 5 to 7 days before and up to 2 weeks after onset of symptoms. Congenitally infected infants may remain infectious for months after birth. Rubella is typically a mild infection in children and is often subclinical in adults. The incubation period ranges from 1 to 21 days.
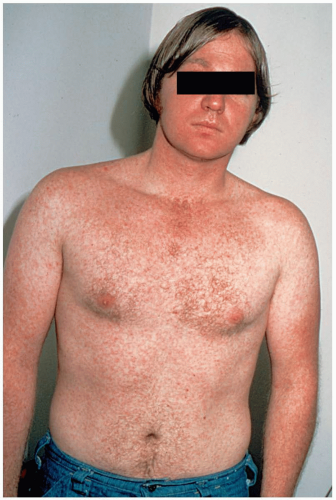 FIGURE 16-7 Rubella. This patient has a viral exanthem. This rash is not distinctive enough to allow a definitive diagnosis based on the clinical presentation alone. (From Goodheart, with permission.) |
Diagnosis
Serum immunoglobulin M (IgM) and acute and convalescent serum IgG levels usually confirm the diagnosis. Rubella virus may be cultured from nasopharyngeal or pharyngeal swabs, urine, blood, and cerebrospinal fluid. Notification to laboratory personnel of possible rubella infection may increase culture sensitivity.
Clinical Complications
Most infections are self-limited, and clinical complications are rare; however, congenital infections are associated with significant morbidity and mortality. Maternal infection in the first trimester results in fetal infection in most cases and congenital defects in 100% of infected infants. In contrast, there is almost no risk of fetal infection or congenital defects after the second trimester.1
Congenital infection may result in spontaneous abortion, intrauterine growth retardation, or stillbirth. Congenital rubella syndrome (CRS) may include mental or physical retardation, hearing loss, cardiac anomalies, ocular anomalies, hepatomegaly and jaundice, purpura, and thrombocytopenia.
Management
Preventative care consists of a two-dose vaccine regimen (part of the measles-mumps-rubella MMR vaccine). The rubella vaccine is a live-attenuated vaccine and is contraindicated in pregnancy. Management of rubella infection generally consists of supportive care, because rubella is usually mild and self-limited. Nonsteroidal antiinflammatory drugs (NSAIDS) are effective for patients with arthralgias.
Postexposure prophylaxis for women exposed early in pregnancy who do not desire pregnancy termination consists of intramuscular immune globulin (20 mL). Consultation with an obstetric or infectious disease specialist (or both) is recommended. Administration of immune globulin within 72 hours after exposure is most effective in preventing infection. There is no effective treatment for infection during pregnancy or for infants with CRS. Contact isolation must be provided for any infant with suspected CRS.
REFERENCES
1. Vander Straten MR, Tyring SK. Rubella. Dermatol Clin 2002;20:225-231.
16-8 Varicella (Chickenpox)
Kimberly J. Center
Clinical Presentation
Varicella (chickenpox) manifests as a characteristic generalized, purpuric, vesicular exanthem occurring about 14 days after infection (incubation period, 10 to 21 days). Lesions appear on the head and trunk and spread centrifugally to cover the body and mucous membranes; the average number of lesions is 300. Lesions begin as red macules that evolve to form clear vesicles on an erythematous base (“dew drop on a rose petal”); over the next 24 to 48 hours, the fluid becomes cloudy and crusting begins. New crops of lesions appear for 1 to 7 days. Typically, lesions in various stages of evolution are evident. Crusts are eventually sloughed, leaving minimal scars, although changes in pigmentation may persist for several weeks. Varicella in vaccinated patients may be less severe, with fewer, nonvesiculopustular lesions and milder systemic symptoms.1 Constitutional symptoms, including fever (100 to 102° F), malaise, anorexia, and mild abdominal pain, occur 24 to 72 hours after the rash appears.
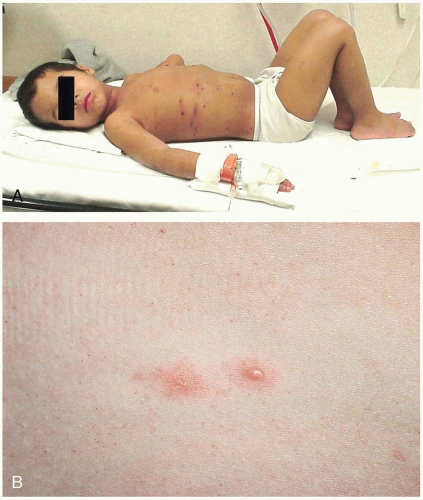 FIGURE 16-8 A: Varicella. Note truncal distribution. (Courtesy of Kimberly J. Center, MD.) B: Varicella. “Dewdrops on rose petals.” (From Goodheart, with permission.) |
Pathophysiology
Varicella-zoster virus (VZV) is the causative agent of chickenpox (primary infection) and zoster (reactivation of latent infection). VZV is highly contagious. Transmission occurs via direct contact with skin lesions or by airborne spread. Respiratory secretions are infectious before the rash appears. The virus remains in the dorsal root ganglia as an inevitable consequence of primary infection, and reactivation results in zoster. After infection, immunity is generally lifelong, and symptomatic reinfection is uncommon in healthy people.
Diagnosis
Diagnostic testing is not required in healthy children. If testing is desired, the direct fluorescent antibody test, polymerase chain reaction testing of a lesion scraping, or viral culture is available.
Clinical Complications
Complications include bacterial superinfection, encephalitis, seizures, cerebellar disease, varicella hepatitis, Reye’s syndrome, thrombocytopenia, and varicella pneumonia (in adolescents and adults).2 Pregnant women may have severe disease, and infection may result in congenital varicella syndrome (cutaneous defects, limb atrophy, microcephaly, mental retardation, seizures, and chorioretinitis).
Management
In healthy children, antiviral therapy is not routinely recommended.2 Antiviral treatment (e.g., acyclovir) is warranted in patients who are older than 12 years of age, have chronic skin or pulmonary conditions, are receiving long-term salicylate therapy, or are taking corticosteroids. Acyclovir should be started within the first 24 to 72 hours of illness.1 A live-attenuated varicella vaccine is recommended for healthy people older than 12 months of age.2
REFERENCES
1. Feder HM Jr, LaRussa P, Steinberg S, Gershon AA. Clinical varicella following varicella vaccination: don’t be fooled. Pediatrics 1997;99:897-899.
2. American Academy of Pediatrics Committee on Infectious Diseases: The use of oral acyclovir in otherwise healthy children with varicella. Pediatrics 1993;91:674-676.
16-9 Pediatric Foreign Body Aspiration
Mark Waltzman
Clinical Presentation
If foreign body aspiration (FBA) in a child is witnessed, the clinical presentation is easily identified as a brief period of choking, gagging, or wheezing, followed by hoarseness, aphonia, or dysphonia. However, up to 50% of patients with FBA do not have a contributing history.1 These patients may present with a variety of respiratory complaints, including cough, dyspnea, and chest pain. FBA should be considered in all pediatric patients who have unilateral lung findings, particularly unilateral wheezing.
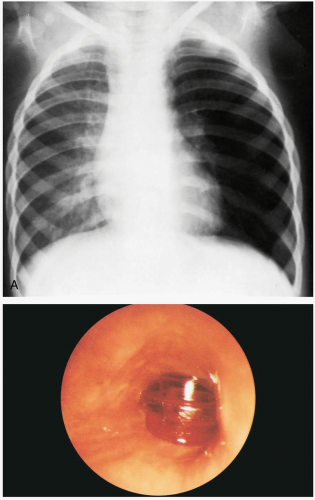 FIGURE 16-9 A: Hyperinflation of the left lung and mediastinal shift to the right. (From Swischuk, with permission.) B: Inhaled plastic foreign body lodged in airway as seen on endoscopy. (From Benjamin, with permission.) |
Pathophysiology
Foreign bodies in the trachea or in a bronchus may lead to air trapping, inflammation, and consolidation. Of special note are complications associated with aspiration of nuts such as peanuts or cashews. The salt and oil may cause an inflammatory response and subsequent pneumonitis.1 In addition, peanuts and similar organic materials may swell, fragment, and crumble, making them difficult to remove.
Diagnosis
Patients may be asymptomatic, initially developing delayed symptoms related to the area of obstruction. Tracheal foreign bodies may produce a “brassy” cough with or without abnormal voice, bidirectional stridor, or complete airway obstruction.2 Bronchial foreign bodies may produce cough, asymmetric wheezing, diminished breath sounds, and hyperresonance to percussion. Because many FBAs are radiolucent, radiographs are useful primarily in detecting indirect signs of a foreign body, such as air trapping, mediastinal shift, atelectasis, or pneumonia.3 Forced expiratory or lateral decubitus chest radiographs may be used in the evaluation of air trapping (the obstructed lung remains inflated during expiration). Absence of physical or radiographic findings does not preclude the possibility of an FBA, and, if clinical suspicion is high, bronchoscopy should be considered.
Clinical Complications
Complications include pneumonia, respiratory distress, and respiratory arrest.4
Management
REFERENCES
1. Cataneo AJ, Reibscheid SM, Ruiz RL Jr, Ferrari GF. Foreign body in the tracheobronchial tree. Clin Pediatr (Phila) 1997;36:701-706.
2. Verghese ST, Hannallah RS. Pediatric otolaryngologic emergencies. Anesthesiol Clin North Am 2001;19:237-256.
3. Rovin JP, Rodgers BM. Pediatric foreign body aspiration. Pediatr Rev 2000;21:86-90.
4. Swanson KL, Prakash UB, Midthun DE, et al. Flexible bronchoscopic management of airway foreign bodies in children. Chest 2002;121:1695-1700.
16-10 Pediatric Pneumonia
Carolyn Trend
Clinical Presentation
Pediatric patients with pneumonia present with a history that is typical of pneumonia. The child is usually acutely ill, with fever, cough, tachypnea, and leukocytosis, although some children present with only mild respiratory symptoms.
Diagnosis
The radiographic appearance of “round pneumonia” and the thymus are relatively unique to pediatrics. “Round pneumonia” is a pulmonary infection that appears as a round, water-density shadow on chest radiograph.1,2 The thymus may produce opacities in the upper lung field that may be evident in very young children. Chest radiography of a “round pneumonia” shows an infiltrate with irregular margins and air bronchograms. The infiltrate, commonly found in the posterior portions of the lungs, usually appears round on frontal projection but may look triangular on lateral views. Some patients have additional parenchymal densities in the ipsilateral or contralateral lung fields. Many patients have hilar enlargement as well.1,2 A child with signs and symptoms consistent with respiratory infection who has radiographic findings consistent with round pneumonia does not require additional diagnostic studies.
 FIGURE 16-10 This well-defined, right mediastinal mass is actually the thymus shadow (arrows). (From Harris and Harris, with permission.) |
The thymus may produce a shadow in bilateral upper lung fields (the so-called “sail sign”) that should not be mistaken for an infiltrate. The thymus is typically symmetric on the anteroposterior or posteroanterior film and is not present in the upper lung fields of the lateral image.
Clinical Complications
Complications of pneumonia include empyema, lung abscess, pneumatocele, respiratory failure, sepsis, and metastatic infection.
Management
Patients with pneumonia should be treated with antibiotics to cover Streptococcus pneumoniae, the most common organism causing round pneumonia.2 Other organisms that may cause infection presenting as round pneumonia include group A streptococci, Klebsiella pneumoniae, and staphylococcal species.
REFERENCES
1. Rose RW, Ward BH. Spherical pneumonias in children simulating pulmonary and mediastinal masses. Radiology 1973;106:179-182.
2. Camargos PA, Ferreira CS. On round pneumonia in children. Pediatr Pulmonol 1995;20:194-195.
16-11 Cystic Fibrosis
Robert Hendrickson
Clinical Presentation
Patients with cystic fibrosis (CF) may present to the emergency department (ED) with infectious, pulmonary, or gastrointestinal manifestations of CF. Patients with CF may have chronic productive cough, obstructive airway disease, sinusitis, and nasal polyps, as well as pulmonary colonization with Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, and Burkholderia cepacia.1 Exacerbations of their pulmonary disease may be obstructive or infectious (e.g., pneumonia, bronchitis). Patients with CF may have chronic pancreatitis, with acute exacerbations presenting as abdominal pain and increased steatorrhea.
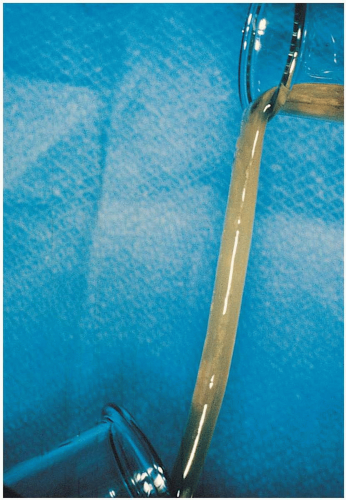 FIGURE 16-11 Duodenal mucus from a patient with cystic fibrosis obtained by means of intraduodenal intubation. Extremely viscid mucus retains its elastic properties even when poured from flask to flask. (From Yamada, with permission.) |
Pathophysiology
CF is an autosomal recessive disorder of chloride channels that is associated with disorders of the pulmonary, gastrointestinal, and urogenital systems.1 A variety of mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7 may lead to an altered chloride channel and CF. Alteration in the chloride channel leads to decreased absorption of water and sodium in epithelial cells (e.g., in the pulmonary tree) and decreased mucociliary clearance.2 This leads to chronic thickening of bronchial secretions and edema, as well as inhibited pulmonary immunity (obstructive and infectious pulmonary disease) and obstruction of seminal vesicles (oligospermia) and the pancreatic duct (pancreatitis).
Diagnosis
Most CF patients seen in the ED already carry the diagnosis of CF. The diagnosis may be made at birth through perinatal screening or in early childhood by recognition of the typical chronic symptoms. The diagnosis may be confirmed by quantitative pilocarpine iontophoresis, or the “sweat test.”2
Clinical Complications
Management
Patients with infectious or obstructive pulmonary exacerbations may require airway control, ventilation, fluid resuscitation, and antibiotics. Obstructive airway symptoms may be treated with inhaled β-adrenergic agonists and anticholinergic agents. Patients with suspected infections should be treated with parenteral antistaphylococcal and antipseudomonal antibiotics.2 Several weeks of antibiotic therapy is required; the specific antibiotic should be based on the patient’s prophylactic regimen and personal and local resistance and should be coordinated with the primary physician. Gastritis or gastroesophageal reflux disease may be treated with protonpump inhibitors. Mild, chronic pancreatitis may require oral pancreatic enzymes. Severe pancreatitis may require bowel rest, pain control, and intravenous hydration.
REFERENCES
1. Rosenstein BJ, Zeitlin PL. Cystic fibrosis. Lancet 1998;351:277-282.
2. Ratjen F, Doring G. Cystic fibrosis. Lancet 2003;361:681-689.
16-12 Kawasaki Disease
Christy Salvaggio
Clinical Presentation
Patients with Kawasaki disease are typically children aged 1 to 2 years who are irritable and have the following symptoms: fever lasting 5 days or longer (high and unresponsive to antipyretics), cervical lymphadenopathy greater than 1.5 cm, nonsuppurative conjunctivitis, mucous membrane lesions (dry, cracked lips; strawberry tongue), hand and/or foot swelling (diffuse erythema/swelling), and an erythematous rash (truncal maculopapular).1,2
Pathophysiology
Kawasaki disease, or “mucocutaneous lymph node syndrome,” is a medium- and large-vessel vasculitis of uncertain origin that has a propensity for coronary arteries.1,2 The cause of Kawasaki disease is most likely multifactorial, including genetic factors (Asian children are at highest risk) as well as seasonal, infectious, and environmental influences.
 FIGURE 16-12 A: “Strawberry tongue.” B: Conjunctivitis of Kawasaki disease. C: Erythema and desquamation may occur in the genital area and on the hands and feet. (A-C, From Goodheart, with permission.) |
Diagnosis
The criteria for Kawasaki disease are fever (5 days or longer) with at least four of the following symptoms: lymphadenopathy, conjunctivitis, mucous membrane involvement, hand/feet erythema or swelling, and an erythematous rash. Children are typically quite irritable and uncomfortable. Children with fever who have fewer than four of the diagnostic criteria may represent “atypical” Kawasaki presentations. Resting tachycardia out of proportion to the fever is common, and most children show some degree of myocarditis and effusion on initial echocardiography. Some children have overt congestive heart failure. Although cardiovascular symptoms predominate, Kawasaki disease may involve multisystem (gastrointestinal, renal, rheumatologic, neurologic) pathology. Symptoms may include vomiting, diarrhea, hepatitis, cholestasis, hydrops of the gallbladder, sterile pyuria, arthritis, arthralgias, and aseptic meningitis. Acute phase reactants, including erythrocyte sedimentation
rate (ESR) and C-reactive protein level (CRP), are elevated, and patients may have a leukocytosis and a cerebrospinal fluid pleocytosis.
rate (ESR) and C-reactive protein level (CRP), are elevated, and patients may have a leukocytosis and a cerebrospinal fluid pleocytosis.
Clinical Complications
Management
A pediatric and/or cardiology consultation should be obtained for any patient with a constellation of symptoms concerning Kawasaki disease, even if not all of the criteria for diagnosis are met.1 Patients with Kawasaki disease may be treated with intravenous immune globulin (IVIG) and acetylsalicylic acid (ASA) therapy. Single-dose IVIG given within 10 days after symptom onset decreases the incidence of coronary artery pathology from 17% to 4%.1 The response to IVIG may vary, but most children are markedly improved and afebrile within 24 hours. The white blood cell count and CRP decrease significantly, but the ESR remains elevated for several weeks after IVIG. Patients may be treated with high-dose aspirin therapy until they are at least 14 days into the illness and have been afebrile for 48 hours, followed by low-dose aspirin until the ESR normalizes and a repeat echocardiogram is normal.1
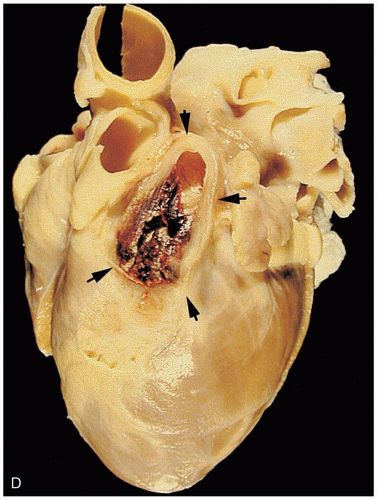 FIGURE 16-12, cont’d D: Coronary artery aneurysm with thrombosis. (From Reece, with permission.) |
REFERENCES
1. Newburger JW, Takahashi M, Burns JC, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med 1986;315:341-347.
2. Rowley AH. Incomplete (atypical) Kawasaki disease. Pediatr Infect Dis J 2002;21:563-565.
16-13 Hand-Foot-and-Mouth Disease
Christy Salvaggio
Clinical Presentation
The large majority of coxsackie infections are asymptomatic. Symptomatic patients with hand-foot-and-mouth disease present with fever, malaise, oral ulcers, and papular cutaneous lesions. The oral lesions are erythematous macules that may evolve into vesicles or ruptured ulcers on the buccal and glossal mucosa and hard palate. Children may complain of a sore throat, refuse to eat, or be drooling secondary to painful oral ulcers.1,2 Cutaneous lesions consist of 3- to 5-mm, tender, gray papules or vesicles with surrounding erythema. The lesions are most concentrated on the distal extremities and the buttocks. Palm and sole involvement is common. The timing of the appearance of cutaneous lesions is variable, and they may precede the appearance of oral ulcers. Oral and cutaneous lesions usually resolve within 1 week.1,2
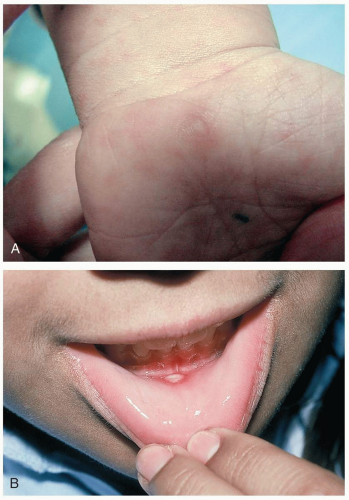 FIGURE 16-13 A: Typical 3- to 7-mm, oval, gray vesicle of the palm. (Courtesy of Christy Salvaggio, MD.) B: Oral lesion of hand-foot-and-mouth disease. Note the oval shape and rim of erythema. (From Goodheart, with permission.) |
Pathophysiology
Hand-foot-and-mouth disease is an enterovirus infection, most commonly caused by coxsackie A16, that manifests as oral mucosal ulcerations and cutaneous papular lesions, especially on the hands and feet.1,3 This is primarily a disease of childhood, usually affecting children younger than 10 years of age. Children younger than 5 years of age are at highest risk. High-risk adults include caretakers and household contacts of infected children. Most enteroviruses are shed in the stool, and hand-foot-and-mouth disease is predominantly spread by the fecal-oral route. Infection rates are high, approaching 100% for close contacts. Hand-foot-and-mouth disease occurs year-round, but infection rates are highest during the summer and fall.1,2,3,4
Diagnosis
Hand-foot-and-mouth disease is essentially a clinical diagnosis. If oral ulcers and classic cutaneous lesions are present, the disease is easy to diagnosis; however, cutaneous lesions may precede any mucosal involvement, making the diagnosis more challenging. Viral cultures or determination of circulating antibodies is rarely needed for diagnosis.1,2 The differential diagnosis for oral ulcers and gingival inflammation includes herpangina, which is also caused by coxsackie viruses, and gingivostomatitis, which is caused by herpes simplex viruses. Neither herpangina nor gingivostomatitis has the cutaneous manifestations classic for hand-foot-and-mouth disease. The oral ulcers of herpangina are more concentrated in the posterior oropharynx. Gingivostomatitis often has significant gingival inflammation, erythema, and tenderness that may precede the appearance of ulcers. Coalescence of ulcers and bleeding mucosa may also be seen. An accurate clinical diagnosis is important, because there is no effective antiviral therapy for hand-foot-and-mouth disease, but acyclovir is effective for herpes gingivostomatitis.1,2
Clinical Complications
Dehydration is the most common complication and may usually be avoided with persistent efforts to encourage oral hydration and adequate pain management. Rarely, hand-foot-and-mouth disease progresses to more serious disease, such as pneumonia, myocarditis, acute flaccid paralysis, encephalitis, or meningitis.1,2,3,4
Management
Although this is usually a self-limited disease, caregivers must be aware that fever may persist for several days and more lesions may appear before symptoms start to resolve. Oral lesions may be painful, and oral hydration
may be difficult. For patients who are moderately to severely symptomatic, topical anesthetics, such as diphenhydramine with or without topical lidocaine, are the mainstay of treatment. A popular combination, called “magic mouthwash,” includes equal parts of 1% viscous lidocaine, diphenhydramine, and aluminum/magnesium hydroxide (e.g., Maalox). Older patients can gargle and spit the solution. For young children, caregivers can paint the solution on the lesions. Because of the potential for lidocaine toxicity, patients should not be prescribed more than 2 mL/kg of the mixed solution (no more than 0.6 mL/kg of 1% lidocaine). Greater quantities may produce seizures or confusion if taken all at once by the child.2,4 Dehydrated children who are refusing liquids should receive intravenous hydration and topical oral anesthetics. Children who continue to refuse liquids may need hospitalization for continued intravenous hydration.4
may be difficult. For patients who are moderately to severely symptomatic, topical anesthetics, such as diphenhydramine with or without topical lidocaine, are the mainstay of treatment. A popular combination, called “magic mouthwash,” includes equal parts of 1% viscous lidocaine, diphenhydramine, and aluminum/magnesium hydroxide (e.g., Maalox). Older patients can gargle and spit the solution. For young children, caregivers can paint the solution on the lesions. Because of the potential for lidocaine toxicity, patients should not be prescribed more than 2 mL/kg of the mixed solution (no more than 0.6 mL/kg of 1% lidocaine). Greater quantities may produce seizures or confusion if taken all at once by the child.2,4 Dehydrated children who are refusing liquids should receive intravenous hydration and topical oral anesthetics. Children who continue to refuse liquids may need hospitalization for continued intravenous hydration.4
16-13 Hand-Foot-and-Mouth Disease
Christy Salvaggio
REFERENCES
1. Stalkup J. Enterovirus infections: a review of clinical presentation, diagnosis, and treatment. Dermatol Clin 2002;20:217.
2. Amir J. Clinical aspects and antiviral therapy in primary herpetic gingivostomatitis. Paediatr Drugs 2001;3:593-597.
3. Glick M. Viral and fungal infections of the oral cavity in immunocompetent patients. Infect Dis Clin North Am 1999;13:817-831.
4. Amir J, Harel L, Smetana Z, et al. Treatment of herpes simplex gingivostomatitis with aciclovir in children: a randomised double blind placebo controlled study. BMJ 1997;314:1800-1803.
16-14 Erythema Infectiosum
Mark Silverberg
Clinical Presentation
Erythema infectiosum (EI) typically afflicts children 5 to 15 years of age, with seasonal peaks in late winter and early spring. It begins with a mild prodrome of low-grade fever and symptoms of an upper respiratory tract infection, soon followed by a symmetric facial rash, giving the “slapped cheek” appearance.1 The rash then spreads to the trunk and extremities in a reticulated pattern. In adults and adolescents, the rash is less common. They are more likely to present after parvovirus B19 infection with arthralgias that are self-limited and resolve within a few weeks.1
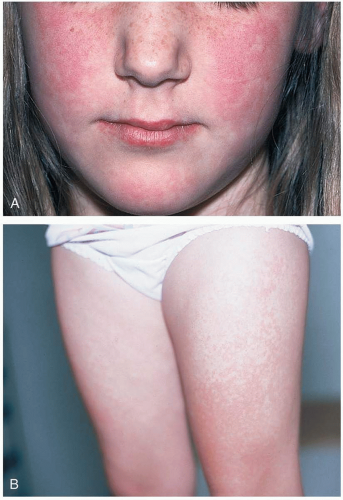 FIGURE 16-14 A: Malar rash of fifth disease (“slapped cheek”). B: Typical reticular pattern on the legs of this patient with fifth disease. (A and B, From Goodheart, with permission.) |
Pathophysiology
EI, also known as “fifth disease,” is an acute infectious illness of childhood caused by parvovirus B19, a nonenveloped, single-stranded DNA virus,2 that results in a characteristic prodrome and rash.1 It is transmitted through respiratory secretion droplets, but infection through blood products has been documented.1,2 Parvovirus B19 has a tendency to attack erythroid precursor cells, especially in individuals who already have chronic hemolytic anemias.1 The distinctive rash of fifth disease is associated with the clearance of viremia and the production of a specific antibody to the virus particles; it is thought to be caused by immune complex deposition.1 In immunocompromised individuals, chronic infection with parvovirus B19 may persist.1,2
Diagnosis
Clinical Complications
Complications may include transient aplastic crisis in hemolytic anemia, arthropathy, myocarditis, encephalitis, idiopathic thrombocytopenic purpura, hemophagocytic syndrome, dermatologic syndromes, rheumatologic diseases, renal disease, and hepatitis.1 Maternal infection during pregnancy usually leads to fetal infection, which has been shown to cause nonimmune hydrops fetalis and fetal death.1 Chronic infection in immunocompromised individuals has brought about chronic anemia.1
Management
The treatment of EI is usually symptomatic. In patients with chronic hemolytic anemias such as sickle cell disease, aplastic crisis may require blood transfusions. Intravenous immune globulin (IVIG) has been successful in treating chronic infection associated with inadequate humoral responses. IVIG has also been used to treat chronic anemia due to B19 infection in immunocompromised individuals.1
REFERENCES
1. Koch WC. Fifth (human parvovirus) and sixth (herpesvirus 6) diseases. Curr Opin Infect Dis 2001;14:343-356.
2. Katta R. Parvovirus B19: a review. Dermatol Clin 2002;20:333-342.
16-15 Henoch-Schönlein Purpura
Adriana Klucar Stoudt
Clinical Presentation
Henoch-Schönlein purpura (HSP) is a self-limited disease that lasts approximately 1 to 2 weeks and has a tendency to recur. The median age at onset is 4 years (range, 6 months to 86 years).1,2,3 The common triad of symptoms consists of purpura, colicky abdominal pain, and arthritis. Hallmark skin findings include symmetric, palpable, nonblanching purpura or petechiae (or both), most commonly on the buttocks and lower extremities. Edema (feet, hands, scalp, and periorbital), gastrointestinal symptoms (colicky abdominal pain, nausea, vomiting, and gastrointestinal bleeding), and muscular complaints (transient lower-extremity arthralgias and arthritis) are common. HSP vasculitis may affect any organ system. Renal involvement may be noted at the time of diagnosis, or it may develop months later. Transient hematuria with or without proteinuria is the most common renal manifestation. Rarely, patients develop hypertension, nephrotic syndrome, rapidly progressive glomerulonephritis, renal insufficiency, or renal failure. Acute scrotal swelling due to inflammation and hemorrhage may occur (2% to 35% of cases). Central nervous system manifestations, including headache, subtle encephalopathy, and seizures, are rare.1 Constitutional symptoms such as malaise and low-grade fever are present in half of patients with HSP. Fever is usually an early sign and is often caused by a precipitating respiratory infection.2
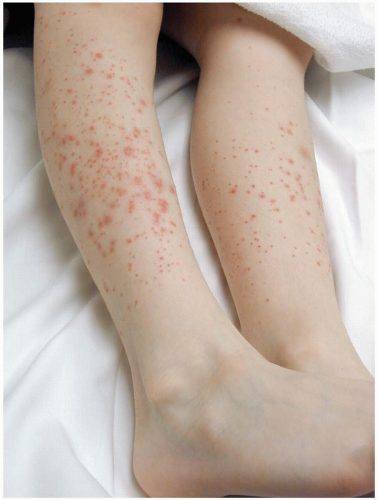 FIGURE 16-15 Lower-extremity purpura in a child with Henoch-Schönlein purpura. (Courtesy of Robert Hendrickson, MD.) |
Pathophysiology
HSP is an immunoglobulin A-mediated vasculitis.
Diagnosis
The diagnosis is made based on recognition of the typical clinical symptoms. In children, palpable purpura with a normal platelet count is adequate to make the diagnosis.1 Routine laboratory findings are typically normal.
Clinical Complications
Approximately 2% of patients develop ileoileal or ileocolic intussusception because of an intestinal wall hematoma lead point.1 Abdominal ultrasonography is the study of choice, because enema studies may miss ileoileal intussusception. Renal complications are rare but may include persistent hypertension and end-stage kidney disease.3
Management
Treatment of HSP is largely supportive. Nonsteroidal antiinflammatory drugs (NSAIDs) may be used to treat joint involvement unless renal insufficiency is present. A short course of corticosteroids may be used, particularly in children with severe abdominal pain, but the efficacy of this treatment is unproven.1 Patients with severe abdominal pain or renal failure should be hospitalized for observation. Mild renal insufficiency may be monitored closely with outpatient urinalysis and serum creatinine values. If the patient does not present with nephritis, serial outpatient urinalysis and serum creatinine levels should be monitored to observe for the development of late-onset renal involvement.1
REFERENCES
1. Szer IS. Henoch-Schonlein purpura: when and how to treat. J Rheumatol 1996;23:1661-1665.
2. Saulsbury FT. Henoch-Schonlein purpura in children. Medicine (Baltimore) 1999;78:395-409.
3. Robson WL, Leung AK. Henoch-Schonlein purpura. Adv Pediatr 1994;41:163-194.
16-16 Juvenile Rheumatoid Arthritis
Leo Ho
Clinical Presentation
Common initial symptoms of juvenile rheumatoid arthritis (JRA) include morning joint stiffness, which usually lasts at least 1 hour, stiffness after periods of inactivity (gel phenomenon), and easy fatigability. Single or multiple joints may be affected. Patients typically present to the emergency department with objective signs of arthritis (joint swelling or limited range of motion accompanied by pain). Oligoarthritis involves fewer than five joints and usually involves the lower extremities (knees, ankles). The mean age at onset is 2 years. Forty percent of patients develop anterior uveitis.1 Polyarthritis involves five or more joints (both large and small joints). Two subtypes exist. Rheumatoid factor (RF)-negative polyarthritis (85%) has a median age at onset at 3 years. RF-positive polyarthritis has a later onset, usually in adolescence.1 Firm, nontender, subcutaneous nodules may develop on extensor skin areas, most commonly the elbow. Systemic-onset disease (10% to 20% of cases) may manifest with arthritis with high-spiking fevers (greater than 39° C), often occurring twice daily for a minimum of 2 weeks. A salmon-pink evanescent rash (2- to 5-mm macules on the trunk and proximal extremities) may be precipitated by scratching or pressure (Koebner’s phenomenon).1
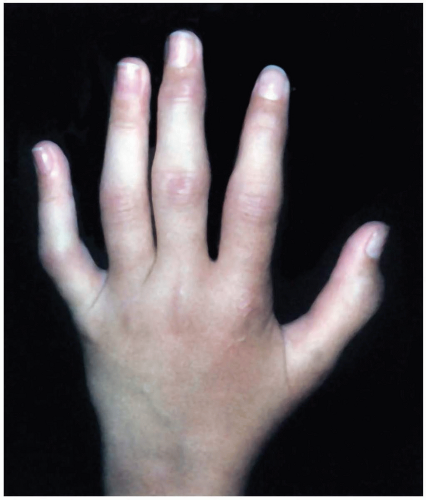 FIGURE 16-16 Hand involvement in systematic juvenile rheumatic arthritis. Note the swelling of multiple joints. (From Staheli, with permission.) |
Pathophysiology
JRA is characterized by synovitis of the peripheral joints and has a strong female predominance. The three principle types of JRA are oligoarthritis (persistent or extended type), polyarticular arthritis, and systemic-onset disease (Still’s disease).1 Less common types include enthesitis arthritis, psoriatic arthritis, and unclassified arthritis.1 The cause of JRA is unknown. However, immunogenetic susceptibility and external triggers may be involved. Possible triggers include viruses (e.g., parvovirus B19, rubella, Epstein-Barr), specific self-antigens (type II collagen), and external trauma.
Diagnosis
Diagnostic criteria include age at onset less than 16 years, oligoarticular or polyarticular arthritis, duration 6 weeks or longer, and exclusion of other forms of juvenile arthritis. Laboratory abnormalities include elevated erythrocyte sedimentation rate and C-reactive protein level, leukocytosis, thrombocytosis, and anemia of chronic disease. Radiographic findings include soft-tissue swelling and periarticular osteopenia adjacent to the involved joint. Late findings include narrowing of the joint space, erosions, subluxations, and ankylosis.
Clinical Complications
Complications include pericarditis, myocarditis, atlantoaxial subluxation, pleural effusions, cricoarytenoid arthritis, and macrophage activation syndrome.
Management
Most cases of oligoarticular JRA respond to nonsteroidal antiinflammatory drugs (NSAIDs), but children with polyarticular disease may require additional medications. Second-line agents include sulfasalazine and methotrexate. Azathioprine and cyclophosphamide are reserved for resistant cases. Intraarticular corticosteroids may be used in some patients, and intravenous corticosteroids are used for systemic illness.1 Physical therapy is crucial for maintaining strength and range of motion.
REFERENCES
1. Woo P, Wedderburn LR. Juvenile chronic arthritis. Lancet 1998; 351:969-973.
16-17 Scarlet Fever
Rakesh D. Mistry
Clinical Presentation
Scarlet fever (SF) manifests abruptly with high fever, severe sore throat, vomiting, chills, and malaise in children. A fine, papular, erythematous rash is initially seen in the flexural areas on the second day of illness. Within 24 hours, the exanthem rapidly becomes generalized and begins to take on a maculopapular, “sandpaper-like” quality, often referred to as scarlatiniform.
Pathophysiology
Infection with group A β-hemolytic streptococcus (GABHS) usually occurs via direct contact or transmission of large nasal droplets. This is followed by an incubation period of 1 to 7 days, during which time the index case often manifests signs of infection. The GABHS organism typically causes direct infection of the affected site—pharynx, skin, or wound. Infecting strains then elicit streptococcal pyrogenic exotoxins A and C, which circulate systemically and produce the characteristic rash and strawberry tongue of scarlet fever.1
 FIGURE 16-17 A: The “sandpaper” rash of scarlet fever. (Courtesy of Mark Silverberg, MD.) B: Scarlet fever. Note skin peeling from the fingertips. (From Goodheart, with permission.) |
Diagnosis
Clinical diagnosis of scarlet fever is based on findings of classic GABHS pharyngitis, accompanied by signs associated with toxin-producing strains, such as “strawberry tongue” and the characteristic exanthem.2 Throat culture is the gold standard of diagnosis. The oropharynx reveals diffuse erythema with palatal petechiae, significant tonsillar enlargement, and purulent exudates of the tonsils or posterior pharynx or both. The rash usually is accentuated in the flexural creases of the neck, elbows, and groin. This is best demonstrated in the antecubital areas, where lines of papules, known as Pastia’s lines, may be seen. The rest of the skin becomes diffusely erythematous; however, the perioral area may be spared, and patients are frequently described as having “circumoral pallor.”1,2,3
Clinical Complications
Complications include otitis media, sinusitis, retropharyngeal or parapharyngeal abscess, cervical adenitis, and sepsis. Poststreptococcal glomerulonephritis may develop despite treatment. A unique complication of scarlet fever is progression to the life-threatening toxic shock syndrome.
Management
Children with scarlet fever require supportive therapy, including hydration, pain control, and antipyresis. Antibiotic therapy may shorten the duration of illness and reduce symptoms; more importantly, it prevents suppurative complications and rheumatic fever. GABHS is susceptible to standard penicillin, which remains the first-line therapy, although studies have documented similar efficacy of easier-dosed penicillins and cephalosporins.2 For penicillin-allergic patients, a course of clindamycin or a macrolide antibiotic is efficacious.1,2,3
REFERENCES
1. Corneli HM. Rapid strep tests in the emergency department: an evidence-based approach. Pediatr Emerg Care 2001;17:272-288.
2. Pichichero ME. Group A streptococcal tonsillopharyngitis: cost-effective diagnosis and treatment. Ann Emerg Med 1995;25:390-403.
3. Kline JA, Runge JW. Streptococcal pharyngitis: a review of pathophysiology, diagnosis, and management. J Emerg Med 1994;12:665-680.
16-18 Rickets
Michael Greenberg
Clinical Presentation
Pathophysiology
Rickets involves inadequate mineralization of bone due to imbalances of phosphorus and calcium metabolism, related to inadequate intake of vitamin D or inadequate exposure to sunlight or both. The poor bone mineralization results in long-bone weakness and inadequate development of bony growth plates.1,2,3 Human milk does not supply vitamin D sufficient to prevent rickets without the concomitant exposure to adequate sunlight.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


