CHAPTER 15 Pain Management
“We must all die. But that I can save (a person) from days of torture, that is what I feel as my great and ever new privilege. Pain is a more terrible lord of mankind than even death itself” (Albert Schweitzer). The treatment and alleviation of pain is a basic human right that exists regardless of age (Yaster et al., 1997; Schechter et al., 2003). The old “wisdom” that young children neither respond to, nor remember, painful experiences to the same degree that adults do is simply untrue (Taddio and Katz, 2005). Many, if not all, of the nerve pathways essential for the transmission and perception of pain are present and functioning by 24 weeks’ gestation (Lee et al., 2005; Lowery et al., 2007). Furthermore, recent research in newborn animals has revealed that the failure to provide analgesia for pain results in “rewiring” the nerve pathways responsible for pain transmission in the dorsal horn of the spinal cord and results in increased pain perception with future painful insults (Fitzgerald and Beggs, 2001; Pattinson and Fitzgerald, 2004). This confirms human newborn research in which the failure to provide anesthesia or analgesia for newborn circumcision resulted not only in short-term physiologic perturbations but also in longer term behavioral changes, particularly during immunization (Taddio et al., 1995; Maxwell et al., 1987; Taddio and Katz, 2005; Anand et al., 2006).
Providing effective analgesia to infants, preverbal children, adolescents, and the mentally and physically disabled poses unique challenges to those who practice pediatric medicine and surgery. In the past, several studies documented that physicians, nurses, and parents underestimate the amount of pain experienced by children and that they overestimate the risks inherent in the drugs used in the treatment of pain (Schechter et al., 1986; Finley et al., 1996; McGrath and Finley, 1996). This is not at all surprising; the guiding principle of medical practice is to do no harm, primum non nocere. Physicians are taught throughout their training that opioids, the analgesics most commonly prescribed in moderate to severe pain, cause respiratory depression, cardiovascular collapse, depressed levels of consciousness, constipation, nausea, vomiting, and, with repeated use, tolerance and addiction. Less potent analgesics, such as nonsteroidal antiinflammatory drugs (NSAIDs), can also cause problems such as bleeding, liver dysfunction, coagulopathies, and impaired wound and bone healing. Thus, physicians at times prescribe insufficiently potent analgesics, recommend inadequate doses, or use pharmacologically irrational dosing regimens because of their overriding concern that children may be harmed by the use of these drugs. The resulting conundrum often results in inadequate treatment for pain and for painful procedures. On the other hand, the adverse effects of pain and the failure to treat it are rarely discussed. In addition to its impact on neurodevelopment, it is known that unrelieved pain interferes with sleep, leads to fatigue and a sense of helplessness, enhances the stress and inflammatory response, and may result in increased morbidity or mortality (Anand et al., 1987).
Nurses may be wary of physicians’ orders (and patients’ requests) as well. The most common prescription order for potent analgesics is “to give as needed” (pro re nata, or PRN). Thus, the patient must know or remember to ask for pain medication, or the nurse must identify when a patient is in pain. These requirements may not always be met by children in pain. Children younger than 3 years of age may be unable to adequately verbalize when or where they hurt. Alternatively, they may be afraid to report their pain. Many children withdraw or deny their pain if pain relief involves yet another terrifying and painful experience—the intramuscular injection or “shot.” Finally, several studies have documented the inability of nurses, physicians, and parents to correctly identify and treat pain, even in postoperative pediatric patients (McGrath and Finley, 1996; Romsing et al., 1996; Fortier et al., 2009).
Societal fears of opioid addiction and lack of advocacy are also causal factors in the under treatment of pediatric pain. Unlike adult patients, pain management in children is often dependent on the ability of parents to recognize and assess pain and on their decision to treat or not treat it (Romsing and Walther-Larsen, 1996; Sutters and Miaskowski, 1997). Even in hospitalized patients, most of the pain that children experience is managed by the patient’s parents (Greenberg et al., 1999; Krane, 2008). Parental misconceptions concerning pain assessment and pain management may therefore result in inadequate pain treatment (Romsing and Walther-Larsen, 1996; Fortier et al., 2009). This is particularly true in patients who are too young or too developmentally handicapped to report their pain themselves. Parents may fail to report pain, either because they are unable to assess it or they are afraid of the consequences of pain therapy. False beliefs about addiction and the proper use of acetaminophen and other analgesics resulted in the failure to provide analgesia to children (Forward et al., 1996; Fortier et al., 2009). In another study, the belief that pain was useful or that repeated doses of analgesics lead to medication not working well resulted in the failure of the parents to provide or ask for prescribed analgesics to treat their children’s pain (Finley et al., 1996). Parental education is therefore essential if children are to be adequately treated for pain. Unfortunately, the ability to educate parents properly about this issue is often limited by insufficient resources, time, and personnel (Greenberg et al., 1999).
Fortunately, the past 25 years have seen an increase in research and interest in pediatric pain management and in the development of pediatric pain services, primarily under the direction of pediatric anesthesiologists (Shapiro et al., 1991; Nelson et al., 2009). Pediatric-pain service teams provide pain management for acute, postoperative, terminal, neuropathic, and chronic pain. This chapter reviews the recent advances in opioid and local anesthetic pharmacology, as well as the various modalities that are useful in the treatment of acute childhood pain.
Pain assessment
The International Association for the Study of Pain (IASP) defines pain as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage” (Merskey et al., 1979). Pain is a subjective experience; operationally it can be defined as “what the patient says hurts” and exists “when the patient says it does hurt.” Infants, preverbal children, and children between the ages of 2 and 7 (Piaget’s Preoperational Thought stage) may be unable to describe their pain or their subjective experiences. This has led many to conclude incorrectly that these children don’t experience pain in the same way as adults. Clearly, children do not have to know (or be able to express) the meaning of an experience in order to have the experience (Anand and Craig, 1996). On the other hand, because pain is essentially a subjective experience, focusing on the child’s perspective of pain is an indispensable facet of pediatric pain management and an essential element in the specialized study of childhood pain. Indeed, pain assessment and management are interdependent and one is essentially useless without the other. The goal of pain assessment is to provide accurate data about the location and intensity of pain, as well as the effectiveness of measures used to alleviate or abolish it.
Multiple validated instruments currently exist to measure and assess pain in children of all ages (von Baeyer and Spagrud, 2007; Crellin et al., 2007; Franck et al., 2000). The sensitivity and specificity of these instruments have been widely debated and have resulted in a plethora of studies to validate their reliability and validity. The most commonly used instruments that measure the quality and intensity of pain are “self-report measures.” In older children and adults, the most commonly used self-report instruments are visual analogue scales (VASs) and numerical rating scales (0 = no pain; 10 = worst pain). However, pain intensity or severity can also be measured in children as young as 3 years of age by using pictures or word descriptors to describe pain. Two common examples include the Oucher Scale (developed by Dr. Judy Beyer), a two-part scale with a vertical numerical scale (0–100) on one side and six photographs of a young child on the other, or the Six-Face Pain Scale, first developed by Dr. Donna Wong and later modified by Bieri et al. (Fig. 15-1) (Beyer and Wells, 1989; Wong and Baker, 1988; Beyer et al., 1990). Because of its simplicity, the Six-Face Pain Scale-Revised is commonly used (Hicks et al., 2001; Bieri et al., 1990). Alternatively, color, word-graphic rating scales, and poker chips have been used to assess the intensity of pain in children. One obvious limitation of all of these self-report measures is their inability to be used in cognitively impaired children or in intubated, sedated, and paralyzed patients.
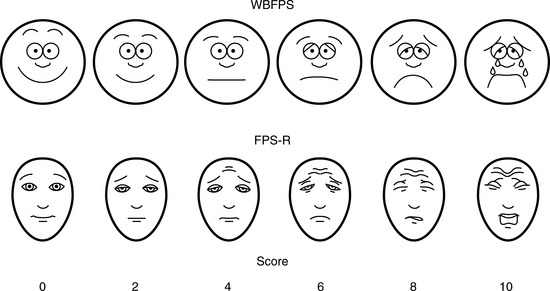
FIGURE 15-1 Six-Face Pain Scale (Top). Original scale developed by Wong et al. Lower, Modified scale by Bieri et al.
(From Bieri D et al.: The Faces Pain Scale for the self-assessment of the severity of pain experienced by children: development, initial validation, and preliminary investigation for ratio scale properties, Pain 41:139, 1990; Beyer JE et al.: Discordance between self-report and behavioral pain measures in children aged 3-7 years after surgery, J Pain Symptom Manage 5:350, 1990.)
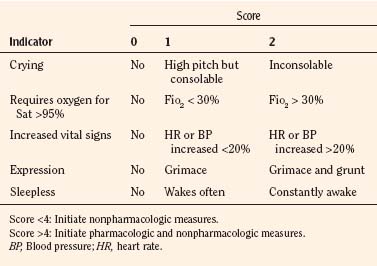
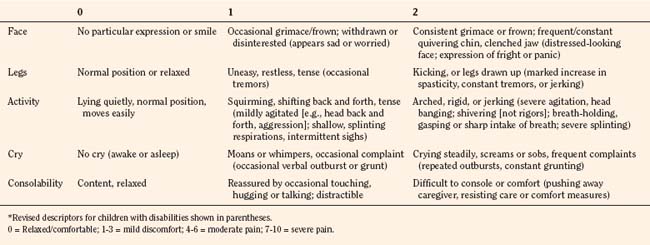
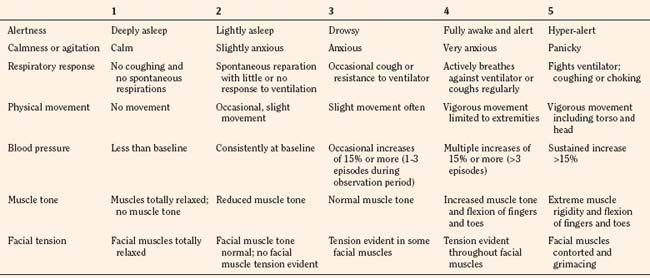
In infants, newborns, and the cognitively impaired, pain has been assessed by measuring physiologic responses to nociceptive stimuli, such as blood pressure and heart rate changes (observational pain scales, OPSs), or by measuring levels of adrenal stress hormones (Krechel and Bildner, 1995). Alternatively, behavioral approaches have used facial expression, body movements, and the intensity and quality of crying as indices of response to nociceptive stimuli. The most appropriate are the Crying, Requires oxygen, Increased vital signs, Expression, and Sleepless (CRIES) score for newborns and the revised Face, Legs, Activity, Cry, and Consolability (FLACC) pain tool for children who have difficulty verbalizing pain (Tables 15-1 and 15-2) (Grunau et al., 1990; Hadjistavropoulos et al., 1994; Voepel-Lewis et al., 2002). Another commonly used pain and sedation tool that uses both behaviors and physiologic parameters is the COMFORT scale, which relies on the measurement of five behavioral variables (alertness, facial tension, muscle tone, agitation, and movement) and three physiologic variables (heart rate, respiration, and blood pressure) (Table 15-3) (Ambuel et al., 1992; Bear and Ward-Smith, 2006). Each is assigned a score ranging from 1 to 5, to give a total score ranging from 8 (deep sedation) to 40 (alert and agitated). A modified COMFORT scale that eliminates physiologic parameters has also been developed (Ista et al., 2005). It is also important to define accurately the location of pain. This is readily accomplished by using either dolls or action figures or by using drawings of body outlines, both front and back. Finally, in the research laboratory, sophisticated new tools such as functional magnetic resonance imaging (MRI) are being used to objectively assess and map pain and its pathways through the central nervous system (CNS) (Tracey and Mantyh, 2007).
Neurophysiology of pain
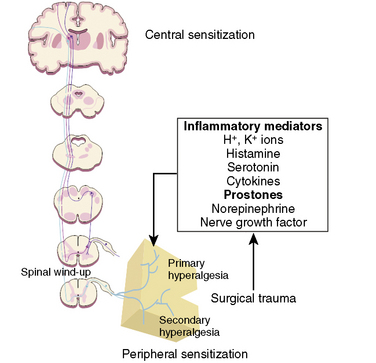
Many if not all of the nerve pathways essential for the transmission, perception, and modulation of pain are present and functioning by 24 weeks of gestation (Fig. 15-2) (Lowry et al., 2007; Lee et al., 2005). Although neural transmission in peripheral nerves is slower in neonates because myelination is incomplete at birth, the major nociceptive neurons in neonates as well as in adults are either unmyelinated C fibers or thinly myelinated Aδ fibers. After an acute injury such as surgical or accidental trauma, inflammatory mediators are released; they lower the pain threshold at the site of injury (primary hyperalgesia) and in the surrounding uninjured tissue (secondary hyperalgesia). These inflammatory mediators, which include hydrogen and potassium ions, histamine, leukotrienes, prostaglandins, cytokines, serotonin (5-HT), bradykinins, and nerve-growth factors make a “sensitizing soup,” which together with repeated stimuli of the nociceptive fibers cause decreased excitatory thresholds and result in peripheral sensitization. They are also targets of therapeutic intervention (Fig. 15-3). Secondary effects of peripheral sensitization include hyperalgesia, the increased response to a noxious stimulus and allodynia, whereby non-nociceptive fibers transmit noxious stimuli resulting in the sensation of pain from non-noxious stimuli.
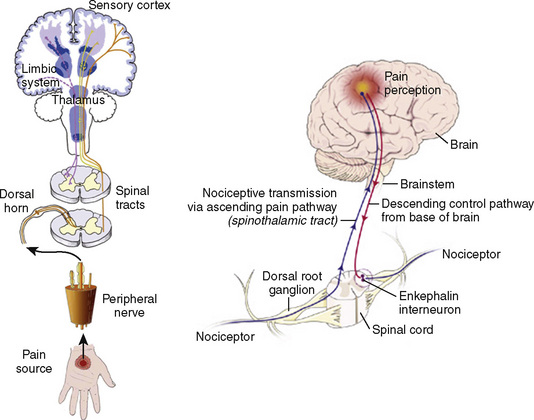
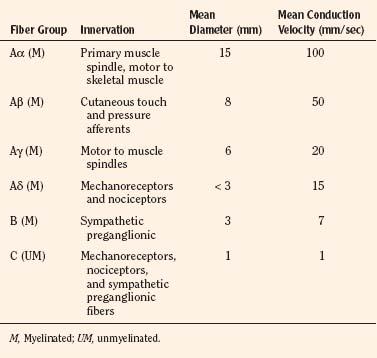
Sensory afferent neurons have a unipolar cell body located in the dorsal root ganglion and are classified by fiber size into three major groups (A, B, C) (Table 15-4). Group A is further subclassified into four subgroups. Sensory fibers that respond to noxious stimulation include small caliber myelinated (Aδ) or fine unmyelinated C fibers. These fibers originate as free nerve endings that can be characterized by their response to specific stimuli such as pressure, heat, and chemical irritants and arise from epidermal and internal receptive fields, including the periosteum, joints, and viscera. The Aδ nociceptors transmit “first pain,” which is well localized, sharp, and lasts as only as long as the original stimulus. The C-fiber, polymodal nociceptors display a slow conduction velocity and respond to mechanothermal and chemical stimuli. This “second pain” is diffuse, persistent, burning, slow to be perceived, and lasts well beyond the termination of the stimulus.
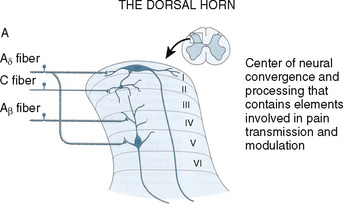
As the primary afferent neurons enter the spinal cord they segregate and occupy a lateral position in the dorsal horn (Fig. 15-4). The Aδ fibers terminate in laminae I, II (substantia gelatinosa), V (nucleus proprius), and X (central canal). The C fibers terminate in laminae I, II, and V, and some enter the dorsal horn through the ventral root. These afferent neurons release one or more excitatory amino acids (e.g., glutamate and aspartate) or peptide neurotransmitters (e.g., substance P, neurokinin A, calcitonin gene-related peptide [CGRP], cholecystokinin, and somatostatin). Second-order neurons that receive these chemical signals integrate the afferent input with facilitatory and inhibitory influences of interneurons and descending neuronal projections. It is this convergence within the dorsal horn that is responsible for much of the processing, amplification, and modulation of pain. Furthermore, the ability to simultaneously process noxious and innocuous stimuli underlies the gate-control theory of pain described by Melzack and Wall (Melzack and Wall, 1965; DeLeo, 2006).
Nociceptive activity in the spinal cord and the ascending spinothalamic, spinoreticular, and spinomesencephalic tracts carry messages to supraspinal centers (e.g., periaqueductal gray, locus coeruleus, hypothalamus, thalamus, and cerebral cortex) where they are modulated and integrated with autonomic, homeostatic, and arousal processes. This modulation, particularly by the endogenous opioids, γ-aminobutyric acid (GABA), and norepinephrine (NE), can either facilitate pain transmission or inhibit it. Modulating pain at peripheral, spinal, and supraspinal sites helps achieve better pain management than targeting only one site and is the underlying principle of treating pain in a multimodal fashion (Fig. 15-3).
Although pain pathways are present at birth, they are often immature (Lee et al., 2005; Lowery et al., 2007). As a result, there are considerable differences in how an infant responds to injury compared with the way an adult does. Within the developing nervous system, inhibitory mechanisms in the dorsal horn of the spinal cord are immature, and inhibition of nociceptive input in the dorsal horn of the spinal cord is less than in the adult. Furthermore, dorsal horn neurons in the newborn have wider receptive fields and lower excitatory thresholds than those in older children (Torsney and Fitzgerald, 2003; Bremner and Fitzgerald, 2008; Fitzgerald and Walker, 2009). Thus, compared with adults, young infants have exaggerated reflex responses to pain. Furthermore, recent research in newborn animals has revealed that the failure to provide analgesia for pain results in “rewiring” the nerve pathways responsible for pain transmission in the dorsal horn of the spinal cord and results in increased pain perception for future painful insults (Fitzgerald and Beggs, 2001; Pattinson and Fitzgerald, 2004). This confirms human newborn research in which the failure to provide anesthesia or analgesia for newborn circumcision resulted not only in short-term physiologic perturbations but also in longer term behavioral changes, particularly during immunization (Maxwell et al., 1987; Taddio et al., 1995; Taddio and Katz, 2005).
Preemptive Analgesia, Preventive Analgesia, and Multimodal Analgesia
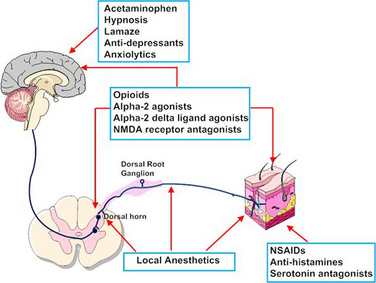
The possibility that pain after surgery might be preemptively prevented or ameliorated by the use of opioids or local anesthetics given preoperatively has been a concept under review (Katz and McCartney, 2002; Moiniche et al., 2002; Oong et al., 2005). Over the past few years the concept of preemptive analgesia has expanded and evolved to include the reduction of nociceptive inputs during and after surgery. This expanded conceptual framework, which includes preoperative, intraoperative, and postoperative analgesia, targets multiple sites along the pain pathway and is referred to as preventive or multimodal analgesia (Ballantyne, 2001; Katz and McCartney, 2002). Indeed, acute pediatric (and adult) pain management is increasingly characterized by a multimodal or “balanced” approach in which smaller doses of opioid and nonopioid analgesics, such NSAIDs, local anesthetics, NMDA antagonists, and α2-adrenergic agonists, are combined to maximize pain control and minimize drug-induced adverse side effects (Fig. 15-5) (DeLeo, 2006). Additionally, a multimodal approach also uses nonpharmacologic complementary and alternative medicine therapies. These alternative medical therapies include distraction, guided imagery, hypnosis, relaxation techniques, biofeedback, transcutaneous nerve stimulation, and acupuncture (Rusy and Weisman, 2000). Taking this approach, activation of peripheral nociceptors can be attenuated with the use of NSAIDs, antihistamines, 5-HT antagonists, and local anesthetics (Fig. 15-5). Within the dorsal horn, nociceptive transmission and processing can be further affected by the administration of local anesthetics, neuraxial opioids, α2-adrenergic agonists (e.g., clonidine and dexmedetomidine), and NMDA receptor antagonists (e.g., ketamine and methadone). Within the CNS, pain can be ameliorated by systemic opioids, α2-agonists, anticonvulsants (e.g., gabapentin and pregabalin), pharmacologic therapies (e.g., benzodiazepines, α2-agonists), and nonpharmacologic therapies (e.g., hypnosis, Lamaze, and acupuncture) that reduce anxiety and induce rest and sleep.
Pharmacologic management of pain: the conundrum of “off-label” drug use
Unfortunately, very few studies have evaluated the pharmacokinetic and pharmacodynamic properties of drugs in children (Conroy and Peden, 2001; Katz and Kelly, 1993). Most pharmacokinetic studies are performed using healthy adult volunteers, adult patients who are only minimally ill, or adult patients in a stable phase of a chronic disease. These data are then extrapolated to infants, children, and adolescents, and the medications are prescribed “off-label.” So little pharmacokinetic and pharmacodynamic testing has been performed in children that they are often considered “therapeutic orphans” (Blumer, 1999). In addition, drug formulations designed for adults are often manipulated and altered by practitioners for use in children (e.g., tablets are dissolved to make a liquid formulation, or suppositories are cut in half). The U.S. Congress has enacted the Best Pharmaceuticals for Children Act, the Pediatric Research Equity Act, and the Food and Drug Administration (FDA) Amendments Act to promote standards and requirements for the use and labeling of pediatric drugs (BPCA, 2002; PREA, 2003; FDAAA, 2007).
Pharmacologic management of pain
Nonopioid Analgesics (or Weaker Analgesics with Antipyretic Activity)
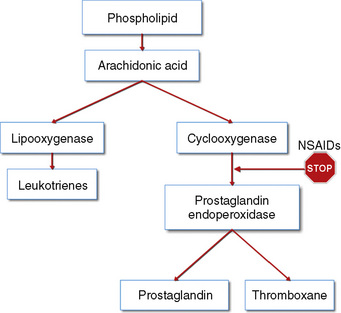
The weaker or milder analgesics with antipyretic activity, of which acetaminophen (paracetamol), salicylate (aspirin), ibuprofen, naproxen, ketoprofen, and diclofenac are common examples, comprise a heterogenous group of NSAIDs and nonopioid analgesics (Table 15-5) (Agency for Health Care Policy and Research, 1992; Yaster, 1997; Tobias, 2000b; Kokki, 2003). They produce their analgesic, antiinflammatory, antiplatelet, and antipyretic effects primarily by blocking peripheral and central prostaglandin and thromboxane production by inhibiting cyclooxygenase (COX) types 1, 2, and 3 (Fig. 15-6). These metabolites of cyclooxygenase sensitize peripheral nerve endings and vasodilate the blood vessels causing pain, erythema, and inflammation.
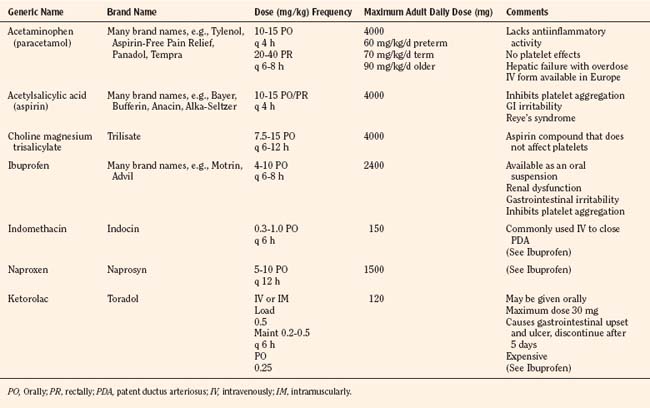
These analgesic agents are administered enterally via the oral or, on occasion, the rectal route and are particularly useful for inflammatory, bony, or rheumatic pain. Parenterally administered agents, such as ketorolac and acetaminophen, are available for use in children in whom the oral or rectal routes of administration are not possible (Murat et al., 2005). Unfortunately, regardless of dose, the nonopioid analgesics are limited by a “ceiling effect” above which pain cannot be relieved by these drugs alone. Because of this, these weaker analgesics are often administered in oral combination forms with opioids such as codeine, oxycodone, or hydrocodone.
Only a few trials have compared the efficacy of these drugs in head to head competition, and in general these studies have shown that there are no major differences in their analgesic effects when appropriate doses of each drug are used. The commonly used NSAIDs, such as ketorolac, diclofenac, ibuprofen, and ketoprofen, have reversible antiplatelet adhesion and aggregation effects that are attributable to the inhibition of thromboxane synthesis (Niemi et al., 1997; Munsterhjelm et al., 2006). As a result, bleeding times are usually slightly increased, but in most instances they remain within normal limits in children with normal coagulation systems. Nevertheless this side effect is of such great concern, particularly in surgical procedures in which even a small amount of bleeding can be catastrophic (e.g., tonsillectomy and neurosurgery), that few clinicians prescribe them even though the evidence supporting increased bleeding is equivocal at best (Moiniche et al., 2003; Cardwell et al., 2005). Finally, many orthopedic surgeons are also concerned about the negative influence of all NSAIDs, both selective and nonselective COX inhibitors, on bone growth and healing (Simon et al., 2002; Einhorn, 2003; Dahners and Mullis, 2004). Thus, most pediatric orthopedic surgeons have recommended that these drugs not be used in their patients in the postoperative period.
The discovery of at least three COX isoenzymes (COX-1, COX-2, and COX-3) has enhanced our knowledge of NSAIDs (Cashman, 1996; Vane and Botting, 1998). The COX isoenzymes share structural and enzymatic similarities, but they are specifically regulated at the molecular level and may be distinguished by their functions. Protective prostaglandins, which preserve the integrity of the stomach lining and maintain normal renal function in a compromised kidney, are synthesized by COX-1 (Vane and Botting, 1998; Moiniche et al., 2003; Levesque et al., 2005). The COX-2 isoenzyme is inducible by proinflammatory cytokines and growth factors, implying a role for COX-2 in both inflammation and control of cell growth. In addition to the induction of COX-2 in inflammatory lesions, it is expressed constitutively in the brain and spinal cord, where it may be involved in nerve transmission, particularly for pain and fever. Prostaglandins made by COX-2 are also important in ovulation and in the birth process (Vane and Botting, 1998; Moiniche et al., 2003; Levesque et al., 2005). The discovery of COX-2 has made possible the design of drugs that reduce inflammation without removing the protective prostaglandins in the stomach and kidneys made by COX-1. In fact, developing a more specific COX-2 inhibitor was a “holy grail” of drug research, because this class of drug was postulated to have all of the desired antiinflammatory and analgesic properties with none of the gastrointestinal and antiplatelet side effects. Unfortunately, the controversy regarding the potential adverse cardiovascular risks of prolonged use of COX-2 inhibitors has dampened much of the enthusiasm for these drugs and has led to the removal of rofecoxib from the market by its manufacturer (Johnsen et al., 2005; Levesque et al., 2005).
Aspirin
Aspirin, one of the oldest and most effective nonopioid analgesics, has been largely abandoned in pediatric practice because of its possible role in Reye’s syndrome, its effects on platelet function, and its gastric irritant properties. Despite these problems, a “sister” compound, choline-magnesium trisalicylate is still prescribed, particularly in the management of postoperative pain and in the child with cancer. Choline-magnesium trisalicylate is a unique aspirin-like compound that does not bind to platelets and therefore has minimal, if any, effects on platelet function (Yaster, 1997). As a result, it can be prescribed to patients with low platelet counts (cancer patients), dysfunctional platelets (uremia), and in the postoperative period. It is a convenient drug to give to children because it is available in both a liquid and tablet form and is administered either twice a day or every 6 hours. However, the association of salicylates with Reye’s syndrome limits its use, even though the risk of developing this syndrome postoperatively is extremely unlikely.
Acetaminophen
The most commonly used nonopioid analgesic in pediatric practice remains acetaminophen, although its analgesic effectiveness in the neonate is unclear (Shah et al., 1998; Anderson, 2008). Unlike aspirin and other NSAIDs, acetaminophen produces analgesia centrally as a COX-3 inhibitor and via activation of descending serotonergic pathways (Graham and Scott, 2005; Anderson, 2008). It is also thought to produce analgesia as a cannabinoid agonist and by antagonizing NMDA and substance P in the spinal cord (Bertolini et al., 2006). Acetaminophen is an antipyretic analgesic with minimal, if any, antiinflammatory and antiplatelet activity and takes about 30 minutes to provide effective analgesia. When administered orally in standard doses, 10 to 15 mg/kg acetaminophen is extremely safe, effective, and has few serious side effects. When administered rectally, higher doses of 25 to 40 mg/kg are required (Rusy et al., 1995; Birmingham et al., 1997). Because of its known association with fulminant hepatic necrosis, the daily maximum acetaminophen dosages, regardless of formulation or route of delivery, in the preterm infant, full-term infant, and older child are 60, 80, and 90 mg/kg respectively (Table 15-5). Thus, when administering acetaminophen rectally it should be given every 8 hours rather than every 4 hours. Finally, an intravenous formulation of acetaminophen is now available in Europe and can be used in patients in whom the enteral route is unavailable. This formulation has been associated with better analgesia than oral acetaminophen in clinical trials in adult patients and is equally effective and less painful than the prodrug formulation of the drug in children (Murat et al., 2005).
Opioids
Overview
Over the past 30 years, multiple opioid receptors and subtypes have been identified and classified. There are 3 primary opioid receptor types, designated mu (µ) (for morphine), kappa (κ), and delta (δ). These receptors are primarily located in the brain and spinal cord, but they also exist peripherally on peripheral nerve cells, immune cells, and other cells (e.g., oocytes) (Sabbe and Yaksh, 1990; Snyder and Pasternak, 2003; Stein and Rosow, 2004). The μ-receptor is further subdivided into several subtypes such as the μ1 (supraspinal analgesia), μ2 (respiratory depression, inhibition of gastrointestinal motility), and μ3 (antiinflammation, leukocytes), which affects the pharmacologic profiles of different opioids (Pasternak, 2001a, 2001b, 2005; Bonnet et al., 2008). Both endogenous and exogenous agonists and antagonists bind to various opioid receptors.
Opioid receptors, which are found anchored to the plasma membrane both presynaptically and postsynaptically, decrease the release of excitatory neurotransmitters from terminals carrying nociceptive stimuli. These receptors belong to the steroid superfamily of G protein–coupled receptors. Their protein structure contains seven transmembrane regions with extracellular loops that confer subtype specificity and intracellular loops that mediate subreceptor phenomena (Stein and Rosow, 2004). These receptors are coupled to guanine nucleotide (GTP)-binding regulatory proteins (G proteins) and regulate transmembrane signaling by regulating adenylate cyclase (and therefore cyclic adenosine monophosphate [cAMP]), various ion channels (K+, Ca,++, Na+) and transport proteins, neuronal nitric oxide synthetase, and phospholipase C and A2 (Fig. 15-7) (Standifer and Pasternak, 1997; Maxwell et al., 2005; Pasternak, 2005). Signal transduction from opioid receptors occurs via bonding to inhibitory G proteins (Gi and Go). Analgesic effects are mediated by decreased neuronal excitability from an inwardly rectifying K+ current, which hyperpolarizes the neuronal membrane, decreases cAMP production, increases nitric oxide synthesis, and increases the production of 12-lipoxygenase metabolites. Indeed, synergism between opioids and NSAIDs occurs as a result of the greater availability of arachidonic acid for metabolism by the 12-lipooxygenase pathway, after blockade of prostaglandin production by NSAIDs (Vaughan et al., 1997). Some of the unwanted side effects of opioids, such as pruritus, may be the result of opioid binding to stimulatory G proteins (Gs) and may be antagonized by low dose infusions of naloxone (Fig. 15-7) (Crain and Shen, 1998, 1996; Maxwell et al., 2005).
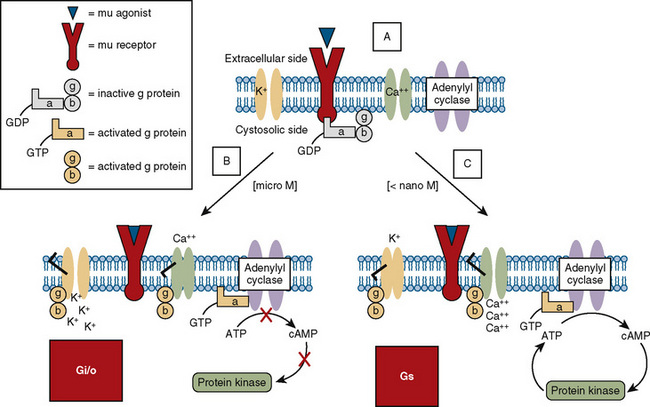
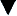 ) (e.g., morphine), it physically associates with the α subunit, causing the latter to release GDP and bind GTP. The GTP binding causes the dissociation of the α subunit from the β-γ subunit and from the receptor. Free α and β-γ subunits are functionally active and directly regulate a number of effector proteins, such as ion channels, adenylyl cyclase, and phospholipase C. B, Classically, the opioid receptor is thought to be a Gi/o coupled receptor. Adenylyl cyclase is inhibited, the potassium channel is open, and the calcium channel is closed. C, At picomolar or nanomolar concentrations, opioid receptors are coupled to Gs proteins. Adenylyl cyclase is activated, the calcium channel is open, and the potassium channel is closed.
) (e.g., morphine), it physically associates with the α subunit, causing the latter to release GDP and bind GTP. The GTP binding causes the dissociation of the α subunit from the β-γ subunit and from the receptor. Free α and β-γ subunits are functionally active and directly regulate a number of effector proteins, such as ion channels, adenylyl cyclase, and phospholipase C. B, Classically, the opioid receptor is thought to be a Gi/o coupled receptor. Adenylyl cyclase is inhibited, the potassium channel is open, and the calcium channel is closed. C, At picomolar or nanomolar concentrations, opioid receptors are coupled to Gs proteins. Adenylyl cyclase is activated, the calcium channel is open, and the potassium channel is closed.Pharmacokinetics
For opioids to effectively relieve or prevent most pain, the agonist must reach the receptor in the CNS. There are essentially two ways that this occurs, either via the bloodstream (after intravenous, intramuscular, oral, nasal, transdermal, or mucosal administration) or by direct application into the cerebrospinal fluid (intrathecal or epidural). Agonists administered via the bloodstream must cross the blood-brain barrier, a lipid membrane interface between the endothelial cells of the brain vasculature and the extracellular fluid of the brain, to reach the receptor. Normally, highly lipid-soluble agonists, such as fentanyl, rapidly diffuse across the blood-brain barrier, whereas agonists with limited lipid solubility, such as morphine, have limited brain uptake. This rule, however, does not hold true for patients of all ages. The blood-brain barrier may be immature at birth and is known to be more permeable in neonates to morphine. Indeed, Kupferberg and Way (1963) demonstrated in a classic paper that morphine concentrations were two to four times greater in the brains of younger rats than older rats despite equal blood concentrations. Spinal administration, either intrathecally (subarachnoid) or epidurally, bypasses the blood and directly places an agonist into the cerebrospinal fluid, which bathes the receptor sites in the spinal cord (substantia gelatinosa) and brain. This “back door” to the receptor significantly reduces the amount of agonist needed to relieve pain and to induce opioid side effects such as pruritus, urinary retention, and respiratory depression (Cousins and Mather, 1984; Sabbe and Yaksh, 1990). After spinal administration, opioids are absorbed by the epidural veins and redistributed to the systemic circulation, where they are metabolized and excreted. Hydrophilic agents such as morphine cross the dura more slowly than more lipid-soluble agents such as fentanyl or meperidine. This physical chemical property is responsible for the more prolonged duration of action of spinal morphine and its very slow onset of action after epidural administration (Sabbe and Yaksh, 1990).
Biotransformation
Effects of Age and Disease
Morphine, meperidine, methadone, codeine, and fentanyl are biotransformed in the liver before excretion by the kidneys. Many of these reactions are catalyzed in the liver by glucuronidation or microsomal mixed-function oxidases that require the cytochrome P450 system, nicotinamide adenine dinucleotide phosphate (NADPH), and oxygen. The cytochrome P450 system is immature at birth and does not reach adult levels of activity until the first month or two of life. The immaturity of this hepatic enzyme system may explain the prolonged clearance or elimination of some opioids in the first few days to weeks of life (Anderson and Lynn, 2009). On the other hand, the P450 system can be induced by various drugs (such as phenobarbital) and substrates, and once the infant is born, it matures regardless of gestational age. Thus, it is the age from birth and not the duration of gestation that determines how premature and full-term infants metabolize drugs.
Morphine is primarily glucuronidated into two forms: an inactive form, morphine-3-glucuronide, and an active form, morphine-6-glucuronide. Both glucuronides are excreted by the kidney. In patients with renal failure, morphine-6-glucuronide can accumulate and cause toxic side effects including respiratory depression (Murtagh et al., 2007; Lotsch, 2005). This is important to consider not only when prescribing morphine but when administering other opioids that are metabolized into morphine, such as codeine.
Distribution and Clearance
The pharmacokinetics of morphine have been extensively studied in adults, older children, and in premature and full-term newborns (Lynn et al., 1991, 1993). After an intravenous bolus, 30% of morphine is protein-bound in the adult vs. only 20% in the newborn. This increase in unbound (“free”) morphine allows a greater proportion of active drug to penetrate the brain. This may in part explain the observation of Way et al. (1965) of increased brain levels of morphine in the newborn and its more profound respiratory depressant effects. The elimination half-life of morphine in adults and older children is 3 to 4 hours and is consistent with its duration of analgesic action. The half-life of elimination (t½β) is more than twice as long in newborns younger than 1 week of age than in older children and adults and is even longer in premature infants (Lynn and Slattery, 1987). Clearance is similarly decreased in the newborn compared with the older child and adult. Thus, infants younger than 1 month of age attain higher serum levels that decline more slowly than older children and adults. This may also account for the increased respiratory depression associated with morphine in this age group. Interestingly, the t½β and clearance of morphine in children older than 2 months of age are similar to adult values; thus, the hesitancy in prescribing and administering morphine in children younger than 1 year of age may not always be warranted. On the other hand, the use of any opioids in children who were born prematurely (fewer than 37 weeks’ gestation) and are less than 52 to 60 weeks’ postconceptional age or who were born at term and are younger than 2 months of age must be restricted to a monitored setting.
Based on its relatively short half-life (3 to 4 hours), one would expect older children and adults to require morphine supplementation every 2 to 3 hours when being treated for pain, particularly if the morphine is administered intravenously. This has led to the use of continuous infusion regimens of morphine and patient-controlled analgesia (see related section) or, alternatively, administration of longer-acting agonists such as methadone. When administered by continuous infusion, the duration of action of opioids that are rapidly redistributed (such as fentanyl, sufentanil, and alfentanil) and are thought to be short-acting may be longer than would be predicted simply by their pharmacokinetics (see Chapter 7, Pharmacology of Pediatric Anesthesia). An exception to this is remifentanil, a μ-opioid receptor agonist with unique pharmacokinetic properties (Burkle et al., 1996).
The pharmacokinetics of remifentanil are characterized by a small volume of distribution, rapid clearances, and low interpatient variability, compared with other intravenous anesthetic and analgesic agents. The drug has a rapid onset of action (the half-time for equilibration between blood and the effect compartment is 1.3 minutes) and a short context-sensitive half-life (3 to 5 minutes) (Bailey, 2002; Welzing and Roth, 2006). This latter property is attributable to hydrolytic metabolism of the compound by nonspecific tissue and plasma esterases. Virtually all (99.8%) of an administered remifentanil dose is eliminated during the half-life of redistribution t½α (0.9 minutes) and the t½β (6.3 minutes). The pharmacokinetics of remifentanil suggest that within 10 minutes of starting an infusion, remifentanil blood levels will have nearly reached steady state. Thus, changing the infusion rate of remifentanil produces rapid changes in drug effect. The rapid metabolism of remifentanil and its small volume of distribution mean that remifentanil does not accumulate. Discontinuing the drug rapidly terminates its effects and has significant intraoperative implications. When remifentanil is administered intraoperatively as part of a balanced or primary opioid general anesthetic, some patients have reportedly awakened in severe pain. This can be because of inadequate loading of a longer-acting opioid, as well as “opioid-induced hyperalgesia,” a paradoxical process by which opioid administration, even for short periods of time, increases the sensitivity to pain and worsens pain when the opioid is discontinued (Crawford et al., 1992; Chu et al., 2008). Finally, remifentanil may be a reasonable alternative to inhaled general anesthetics in newborn infants undergoing surgery, because it only briefly interferes with the control of breathing and because this effect is terminated shortly after discontinuing the drug (Davis et al., 2001; Galinkin et al., 2001).
Commonly Used Oral Opioids
Codeine, oxycodone (the opioid in Tylox and Percocet), and hydrocodone (the opioid in Vicodin and Lortab) are opioids that are commonly used to treat pain in children and adults, and are quite useful when making the transition from parenteral to enteral analgesia (Table 15-6). Methadone and sustained release formulations of morphine, oxycodone, oxymorphone, and hydromorphone are commonly used to treat chronic medical pain (e.g., cancer) and in postoperative surgical or trauma patients with long recuperative times (e.g., pectus excavatum or posterior spine surgery). Codeine, oxycodone, and hydrocodone are most commonly administered in the oral form, often in combination with acetaminophen. Although the combination drugs are convenient, and acetaminophen potentiates the analgesia produced by codeine—allowing the practitioner to use less opioid to achieve satisfactory analgesia—the combination of drugs significantly increases the risk of acetaminophen toxicity. Acetaminophen toxicity may result from a single toxic dose, from repeated ingestion of large doses of acetaminophen (e.g., in adults 7.5 to 10 g/day for 1 to 2 days, in children 60 to 420 mg/kg per day for 1 to 42 days), or from chronic ingestion. However, hepatotoxicity can also occur inadvertently when patients with poorly controlled pain increase the number of combination tablets they need to control their pain or when they are receiving more than one source of acetaminophen (Heubi et al., 1998). The latter occurs because so many prescription and over-the-counter drug products contain acetaminophen (e.g., cold remedies). Because of this risk, the preferred method is to prescribe opioids and acetaminophen (or ibuprofen) separately.
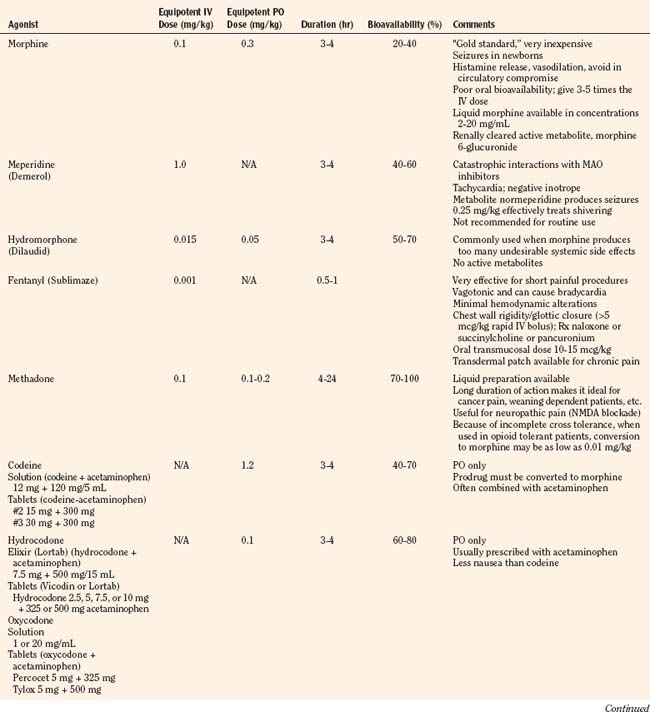
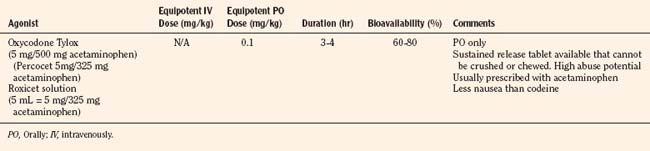
In equipotent doses, oral analgesics have similar effects and side effects including analgesia, sedation, cough suppression, pruritus, nausea, vomiting, constipation and respiratory depression (Table 15-6). Yet the responses of patients to individual opioids can vary markedly, even among these μ-opioid agonists (Pasternak, 2001a, 2005). Understanding this variability greatly enhances the ability to treat patients appropriately. Codeine is a case in point; although readily available, it is very nauseating, and many patients claim they are allergic to it because it so commonly induces vomiting. On the other hand, some differences may be more based on folklore rather than reality. Many physicians falsely believe that meperidine has less of an effect on the sphincter of Oddi than other opioids and therefore prescribe it for patients with gallbladder disease. In fact, meperidine offers no advantage over other opioids and has a serious disadvantage, namely catastrophic interactions with monoamine oxidase (MAO) inhibitors.
Patient-Controlled Analgesia
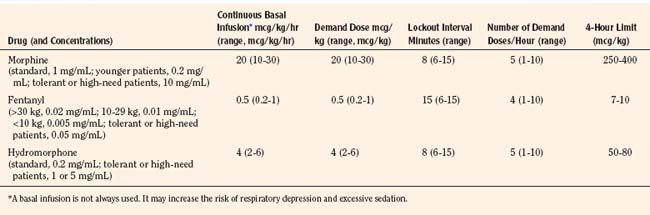
Historically, pain medications have been administered on a demand or PRN basis (Krane, 2008). When drugs are given PRN, the patient or caregiver must recognize that pain exists, summon a nurse and await the preparation and administration of the analgesic. Even in the best of circumstances, there is a delay between the patient’s request and the provider’s response (Krane, 2008). Around-the-clock administration of analgesics at intervals based on population pharmacokinetics (e.g., every 4 hours) is not always effective because there are enormous individual variations in pain perception and opioid metabolism. Knowledge of opioid pharmacokinetics suggests that intravenous boluses of intermediate-acting opioids such as morphine may be needed as often as every 1 to 2 hours in order to avoid marked fluctuations in plasma drug levels, but generally they are ordered no more frequently than every 4 hours. One way to achieve this goal is via continuous intravenous opioid infusions. This approach provides steady analgesic levels and has been used with great safety and efficacy in children; however, because neither the perception nor the intensity of pain is constant, continuous opioid infusions do not adequately treat pain in all patients (Lynn et al., 2000). For example, a postoperative patient may be very comfortable resting in bed and may require little adjustment in opioid dosing. This same patient may experience excruciating pain when coughing, voiding, or getting out of bed. Receiving the same dose of opioid in both instances may result in either oversedation or under treatment. Thus, rational pain management requires some form of titration to effect whenever any opioid is administered. In order to give patients some measure of control over their pain therapy, analgesia on demand or patient-controlled analgesia (PCA) devices were developed (Berde et al., 1991; Yaster et al., 1997). These are microprocessor driven pumps that the patient controls to self-administer intermittent, predetermined, small doses of opioid whenever a need for more pain relief is felt. The opioid, usually morphine, hydromorphone, or fentanyl is administered either intravenously or subcutaneously (Table 15-7). The dosage of opioid, number of demand doses (“boluses”) per hour, and the time interval between boluses (the “lock-out period”) are programmed into the equipment by the pain-service physician to allow maximum patient flexibility and sense of control with minimal risk of overdosage. Generally, because older patients know that if they have severe pain they can obtain relief immediately, many prefer dosing regimens that result in mild to moderate pain in exchange for fewer side effects such as nausea or pruritus. Morphine is the most commonly prescribed opioid. Typically it is prescribed at a bolus dose of 20 mcg/kg, at a rate of up to 5 boluses/hour, and with a lock-out interval between each bolus of 6 to 8 minutes (Table 15-7). Variations include larger boluses (30 to 50 mcg/kg) and shorter time intervals (5 minutes).






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


