Molecular Biology of Brain Injury
Patrick M. Kochanek
Hülya Bayir
Larry W. Jenkins
Robert S.B. Clark
 The two distinct types of secondary injury are (a) evolution of damage mediated by endogenous secondary injury cascades and (b) damage that results from secondary insults (e.g., hypotension) in the field, emergency department, or ICU.
The two distinct types of secondary injury are (a) evolution of damage mediated by endogenous secondary injury cascades and (b) damage that results from secondary insults (e.g., hypotension) in the field, emergency department, or ICU. Ischemia is an important injury mechanism that plays varying roles across the spectrum of PICU-relevant central nervous system (CNS) insults.
Ischemia is an important injury mechanism that plays varying roles across the spectrum of PICU-relevant central nervous system (CNS) insults. Cell death pathways after CNS injury include necrosis, apoptosis, autophagy, and necroptosis. Some cells display mixed phenotypes with characteristics of two or more of these pathways.
Cell death pathways after CNS injury include necrosis, apoptosis, autophagy, and necroptosis. Some cells display mixed phenotypes with characteristics of two or more of these pathways. Key initiators of damage in the evolution of secondary injury include energy failure, excitotoxicity, oxidative stress, and trophic factor withdrawal.
Key initiators of damage in the evolution of secondary injury include energy failure, excitotoxicity, oxidative stress, and trophic factor withdrawal. Apoptosis after CNS injury can result from activation of a number of mechanisms, including (a) an intrinsic (mitochondrial) pathway, (b) an apoptosis-inducing factor pathway, (c) an endoplasmic reticulum stress pathway, and (d) an extrinsic (extracellular signaling) pathway. Agerelated differences in the cell death response to brain injury could be highly important.
Apoptosis after CNS injury can result from activation of a number of mechanisms, including (a) an intrinsic (mitochondrial) pathway, (b) an apoptosis-inducing factor pathway, (c) an endoplasmic reticulum stress pathway, and (d) an extrinsic (extracellular signaling) pathway. Agerelated differences in the cell death response to brain injury could be highly important. An endogenous neuroprotectant response to CNS injury occurs and includes a variety of pathways: adenosine, heat shock proteins, and a variety of antiapoptotic proteins such as Bcl-2.
An endogenous neuroprotectant response to CNS injury occurs and includes a variety of pathways: adenosine, heat shock proteins, and a variety of antiapoptotic proteins such as Bcl-2. Brain swelling after CNS injury results from cellular swelling, vasogenic edema, osmolar swelling, mixed edema patterns, and/or an increase in cerebral blood volume.
Brain swelling after CNS injury results from cellular swelling, vasogenic edema, osmolar swelling, mixed edema patterns, and/or an increase in cerebral blood volume. Inflammation after CNS injury plays an important role in exacerbation of secondary damage and in initiating signals for repair and regeneration.
Inflammation after CNS injury plays an important role in exacerbation of secondary damage and in initiating signals for repair and regeneration. Axonal injury involves a process that includes secondary axotomy after axonal stretch and/or ischemia.
Axonal injury involves a process that includes secondary axotomy after axonal stretch and/or ischemia. Key secondary extracerebral insults that adversely impact outcome across CNS injury etiologies include hypotension, hypoxemia, hyperthermia, and hypoglycemia/hyperglycemia.
Key secondary extracerebral insults that adversely impact outcome across CNS injury etiologies include hypotension, hypoxemia, hyperthermia, and hypoglycemia/hyperglycemia.Knowledge of the pathophysiology of central nervous system (CNS) injury, such as intracranial and cerebrovascular dynamics, has guided brain-oriented therapy since the inception of critical care medicine. In the chapters of Section III, the pathophysiology of each disease process is described, along with how it influences outcome and guides therapy. Although therapy that is based solely on physiologic parameters, such as intracranial pressure (ICP) and cerebral blood flow (CBF), is important, it has limitations. For example, some patients with well-controlled cerebral hemodynamics still have poor outcome. Indeed, some recent studies in the field of neurocritical care have questioned the relative importance of control of raised ICP to outcome (1). For example, the standard medical care group had significantly better long-term outcome in the recent randomized controlled trial (RCT) of decompressive craniectomy in adults with severe traumatic brain injury (TBI) despite substantially better control of ICP in the surgery group. Many explanations have been suggested to explain this finding, and the authors of this chapter do not suggest discounting the importance of ICP in severe TBI; however, one possibility is that the rapid weaning of other neurocritical care therapies such as osmolar therapy, sedation, barbiturates, etc., led to a relative deficit in therapies targeting other mechanism of secondary injury in brain such as excitotoxicity in the patients treated with decompression. We, thus, are working in an exciting era in which knowledge of the pathobiology of CNS injury is gaining importance for the intensivist and can augment pathophysiology-based therapy. Insight into secondary brain injury at the biochemical, cellular, and molecular levels has finally begun to yield dividends, with efficacy shown for several new therapies in ICU-relevant CNS injuries (2,3,4,5). This chapter builds on the material in the preceding chapter that addresses developmental issues of the CNS relevant to critical care. Where possible, this information on the bedside care of critically ill patients is linked with clinical data. It is hoped that this chapter and its counterpart (Chapter 55) aptly set the stage for the following chapters that discuss the individual CNS disorders encountered in the practice of pediatric critical care medicine.
INJURY RESPONSE IN THE IMMATURE BRAIN
Most research on developmental brain injury suggests that the immature brain is more resistant to injury and more capable of recovery than is the adult brain due to enhanced plasticity (ability to rewire or repair). For example, despite similar impact parameters, the immature brain exhibits less neuronal death than the adult brain in experimental TBI. Similarly, tolerance of the immature brain to status epilepticus (SE) is greater than in the adult in experimental models (6). Classic studies of ablative brain lesions in laboratory models, such as hemispherectomy in cats, show that the earlier the brain lesion occurs in development, the better the outcome (7). Unfortunately, critical windows in development represent periods of the brain’s enhanced vulnerability to specific developmental
processes (8) (see Chapter 55). Some developmental aspects predispose the immature brain to greater injury than the adult. For example, the small size of the infant skull may predispose to a more diffuse pattern of injury from a traumatic impact than does the adult skull (9). An injury that is more global than focal may yield a more profound disruption of function. Much of the controversy on this point results from the fact that outcomes from brain injury in infants and young children are often poorer than in adults (10). For example, outcome from out-of-hospital cardiac arrest is worse in children than in adults. However, the mechanism that underlies the cardiac arrest in children—asphyxia—is particularly injurious to the brain, compared with ventricular fibrillation (VF) (11). Similarly, reported outcomes in infants from severe TBI can be as bad as or worse than those in adults (10). This finding likely results from the contribution of abusive head trauma (AHT) to severe TBI in infancy. Thus, although most data suggest that the developing brain exhibits both resistance and resilience to injury versus the adult, the mechanisms of injury are often more severe in children. Our challenges for developing new therapies are no less than in adults.
processes (8) (see Chapter 55). Some developmental aspects predispose the immature brain to greater injury than the adult. For example, the small size of the infant skull may predispose to a more diffuse pattern of injury from a traumatic impact than does the adult skull (9). An injury that is more global than focal may yield a more profound disruption of function. Much of the controversy on this point results from the fact that outcomes from brain injury in infants and young children are often poorer than in adults (10). For example, outcome from out-of-hospital cardiac arrest is worse in children than in adults. However, the mechanism that underlies the cardiac arrest in children—asphyxia—is particularly injurious to the brain, compared with ventricular fibrillation (VF) (11). Similarly, reported outcomes in infants from severe TBI can be as bad as or worse than those in adults (10). This finding likely results from the contribution of abusive head trauma (AHT) to severe TBI in infancy. Thus, although most data suggest that the developing brain exhibits both resistance and resilience to injury versus the adult, the mechanisms of injury are often more severe in children. Our challenges for developing new therapies are no less than in adults.
PATHWAYS AND APPLICATIONS TO PEDIATRIC INTENSIVE CARE
Secondary Injury
The essence of neurointensive care is the prevention of secondary injury. Two distinct types of secondary injury are  frequently lumped into a single concept. First, endogenous secondary injury cascades evolve in the minutes to days after the initial insult. Processes such as excitotoxicity, oxidative stress, and delayed neuronal death cascades kill neurons and injure other components of the CNS at varying rates. Emerging evidence supports the concept that, in some cases, these cascades may be able to be abrogated, resulting in salvage of injured tissue and improved recovery of function. Efficacy in large RCTs has been reported for mild hypothermia after both cardiac arrest from VF in adults and perinatal asphyxia (2,3,5). Similarly, thrombolytics have shown efficacy in large, randomized, controlled clinical trials in stroke in adults (4). Although these therapies are being further evaluated and only beginning to be integrated into standard care, it is likely that additional therapies will follow. This chapter is focused on those events in injury evolution that may represent therapeutic targets.
frequently lumped into a single concept. First, endogenous secondary injury cascades evolve in the minutes to days after the initial insult. Processes such as excitotoxicity, oxidative stress, and delayed neuronal death cascades kill neurons and injure other components of the CNS at varying rates. Emerging evidence supports the concept that, in some cases, these cascades may be able to be abrogated, resulting in salvage of injured tissue and improved recovery of function. Efficacy in large RCTs has been reported for mild hypothermia after both cardiac arrest from VF in adults and perinatal asphyxia (2,3,5). Similarly, thrombolytics have shown efficacy in large, randomized, controlled clinical trials in stroke in adults (4). Although these therapies are being further evaluated and only beginning to be integrated into standard care, it is likely that additional therapies will follow. This chapter is focused on those events in injury evolution that may represent therapeutic targets.
frequently lumped into a single concept. First, endogenous secondary injury cascades evolve in the minutes to days after the initial insult. Processes such as excitotoxicity, oxidative stress, and delayed neuronal death cascades kill neurons and injure other components of the CNS at varying rates. Emerging evidence supports the concept that, in some cases, these cascades may be able to be abrogated, resulting in salvage of injured tissue and improved recovery of function. Efficacy in large RCTs has been reported for mild hypothermia after both cardiac arrest from VF in adults and perinatal asphyxia (2,3,5). Similarly, thrombolytics have shown efficacy in large, randomized, controlled clinical trials in stroke in adults (4). Although these therapies are being further evaluated and only beginning to be integrated into standard care, it is likely that additional therapies will follow. This chapter is focused on those events in injury evolution that may represent therapeutic targets.In contrast to the endogenous cascades, a parallel form of secondary injury involves critically ill patients who suffer secondary insults in the field, emergency room, or ICU. These secondary insults produce adverse consequences on a CNS with enhanced vulnerability after cardiac arrest, stroke, TBI, SE, or CNS infection. For example, hypoxemia, at a level tolerated by the normal brain, can have detrimental effects after TBI. (Why the injured brain is highly vulnerable to secondary insults is discussed later.) Thus, the biochemical, cellular, and molecular aspects of endogenous damage evolution are discussed, followed by a discussion of the mechanisms that underlie enhanced vulnerability of the injured brain to secondary insults in the PICU.
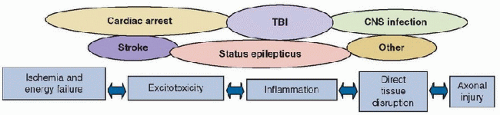 FIGURE 56.1. Overview of the major pathways of secondary injury after PICU-relevant insults to the CNS. Five categories of injury include ischemia and energy failure, excitotoxicity, inflammation, direct tissue disruption, and axonal injury. |
Evolution of Secondary Injury
Presenting a complete synthesis of the mechanisms involved in the evolution of secondary damage after CNS injury is challenging, particularly when one considers that the concepts discussed in this chapter serve as the basis for all PICU-relevant CNS insults. It is necessary to consider studies in models of CNS injury and, where possible, the relevant clinical conditions. We will focus on global cerebral ischemia/cardiac arrest, focal cerebral ischemia/stroke, and TBI. Where possible, insight into the relevance of these mechanisms in epilepsy and CNS infection will be included.
Studies have begun to unravel those mechanisms producing secondary damage that are relevant to cardiac arrest, stroke, TBI, SE, CNS infection, and other insults. Five categories of mechanisms can be defined (Fig. 56.1): those associated with (a) ischemia and energy failure and (b) excitotoxicity, both initiating cell death cascades; (c) inflammation; (d) cerebral swelling; and (e) axonal injury. Within each category, a constellation of mediators of secondary damage is involved (12). The quantitative contribution of each mediator to outcome and the interplay between these mediators remains unclear and varies with the insult. A sixth component of the response to brain injury will be briefly discussed, namely, the role of endogenous neuroprotectants, repair, and regeneration. The ultimate result of the primary injury from TBI, which sets into motion these six cascade initiators, is summarized in Figure 56.2. The details of these initiators represent the crux of information to follow.
Study of the acute biochemical and molecular aspects of human brain injury has been limited, particularly in infants and children; however, several methods have been used, most commonly in TBI and occasionally in asphyxia. These include (a) the analysis of brain biochemistry and molecular biology using ventricular cerebrospinal fluid (CSF); (b) assessment of brain interstitial fluid by cerebral microdialysis; (c) imagingrelated tools, such as positron emission tomography (PET) and MR spectroscopy; and (d) the assessment of molecular markers in brain tissue obtained from patients treated with surgical decompression. Assessment of postmortem brain tissue has provided molecular clues in some cases. We will discuss these clinical studies supporting proposed mechanisms whenever possible.
Ischemia
Global Cerebral Ischemia. The brain is exquisitely vulnerable to ischemia, which represents a unifying mechanism involved in the evolution of secondary damage across CNS  insults. When CBF globally ceases, such as in cardiac arrest,
insults. When CBF globally ceases, such as in cardiac arrest,
a stereotypic time course of events occurs that is initiated by acute cellular energy failure. These changes are outlined in Figure 56.3. Phosphocreatine is depleted in 1 minute, and the adenylate energy charge is depleted in ˜5 minutes. After this time, no useful energy can be made available to ATP-requiring reactions (13). Membrane failure follows, with loss of ionic gradients, increases in intracellular calcium (Ca2+) and sodium (Na+), and a decrease in intracellular potassium (K+). Free
fatty acid release from the neuronal membrane also occurs, and electroencephalogram (EEG) and evoked potentials fail. If energy failure is sustained beyond a critical period, an irreversible insult to neurons occurs and is believed to be manifest by acute necrotic death, with membrane failure, cellular Na+ and Ca2+ accumulation, cellular swelling, and acute failure of organelles, such as mitochondria. However, during complete global brain ischemia, assessment of brain tissue with light microscopy reveals no obvious pathologic derangement, and electron microscopy reveals only chromatin clumping (14). The resultant damage to neurons, glia, and white matter does not manifest until reperfusion, suggesting a critical role for reperfusion in the evolution of secondary damage, as well as the intriguing possibility that even prolonged periods of ischemia might be survived with good outcome if the deleterious consequences of reperfusion could be eliminated. At this writing, the duration of irreversibility for global cerebral ischemia in VF cardiac arrest is believed to be between 5 and 10 minutes, although it can be modified by factors that include temperature, blood glucose concentration, and pH, among others. Although these principles generally hold true for cardiac arrest that results from asphyxia (the most common form in infants and children), some aspects are unique. A more comprehensive discussion of the specific pathophysiology of asphyxia is discussed in Chapter 65.
insults. When CBF globally ceases, such as in cardiac arrest, a stereotypic time course of events occurs that is initiated by acute cellular energy failure. These changes are outlined in Figure 56.3. Phosphocreatine is depleted in 1 minute, and the adenylate energy charge is depleted in ˜5 minutes. After this time, no useful energy can be made available to ATP-requiring reactions (13). Membrane failure follows, with loss of ionic gradients, increases in intracellular calcium (Ca2+) and sodium (Na+), and a decrease in intracellular potassium (K+). Free
fatty acid release from the neuronal membrane also occurs, and electroencephalogram (EEG) and evoked potentials fail. If energy failure is sustained beyond a critical period, an irreversible insult to neurons occurs and is believed to be manifest by acute necrotic death, with membrane failure, cellular Na+ and Ca2+ accumulation, cellular swelling, and acute failure of organelles, such as mitochondria. However, during complete global brain ischemia, assessment of brain tissue with light microscopy reveals no obvious pathologic derangement, and electron microscopy reveals only chromatin clumping (14). The resultant damage to neurons, glia, and white matter does not manifest until reperfusion, suggesting a critical role for reperfusion in the evolution of secondary damage, as well as the intriguing possibility that even prolonged periods of ischemia might be survived with good outcome if the deleterious consequences of reperfusion could be eliminated. At this writing, the duration of irreversibility for global cerebral ischemia in VF cardiac arrest is believed to be between 5 and 10 minutes, although it can be modified by factors that include temperature, blood glucose concentration, and pH, among others. Although these principles generally hold true for cardiac arrest that results from asphyxia (the most common form in infants and children), some aspects are unique. A more comprehensive discussion of the specific pathophysiology of asphyxia is discussed in Chapter 65.
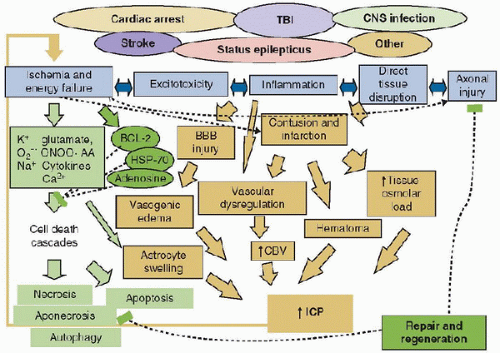 FIGURE 56.2. Secondary cascades triggered by the five initiators of secondary damage identified in Figure 56.1 are shown for cell death cascades (gray), and for brain swelling-related process (stippled gray). In addition, a sixth category of endogenous neuroprotectants and repair and regeneration is initiated; the components of this cascade are shown in black. TBI, traumatic brain injury; O2–, superoxide anion; ONOO, peroxynitrite; AA, arachidonic acid; HSP-70, heat shock protein 70; ICP, intracranial pressure. |
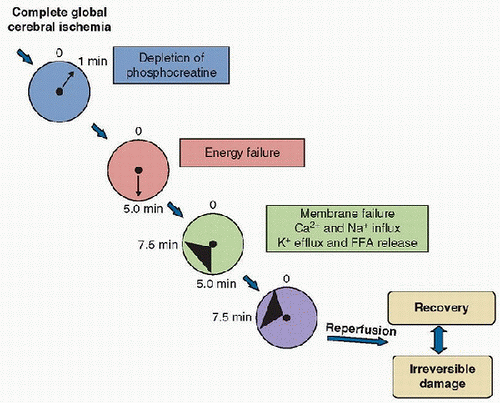 FIGURE 56.3. Temporal sequence of events that occurs in global cerebral ischemia, as seen with a “square-wave” ischemic brain insult, such as VF cardiac arrest. Although ischemia rapidly sets the stage for damage, histology—even by electron microscopy—reveals little damage during the period of ischemia. With ischemic durations beyond 10 minutes at normothermia, considerable damage is manifest with reperfusion. FFA, free fatty acid. |
Focal Cerebral Ischemia. Unlike global brain ischemia, focal ischemic insults (strokes) produce brain regions with different degrees of blood flow reduction related to the vascular anatomy surrounding the area of occlusion. The typical result is an ischemic core, in which flow reductions are profound, surrounded by penumbral brain regions that have less severe but still compromising CBF reductions. The consequences of penumbral CBF reductions on brain metabolism have been reviewed (15). The most sensitive biochemical or molecular event to CBF reduction in the ischemic penumbra is protein synthesis, which is inhibited by ˜50% at a CBF of 55 mL/100 g/min and generally fails below 35 mL/100 g/min (Fig. 56.4). Other homeostatic processes that fail at decreasing CBF thresholds in focal ischemia include glucose utilization below 25 mL/100 g/min and ATP depletion, beginning at a CBF less than ˜20 mL/100 g/min. Peri-infarct depolarization waves occur at CBF levels of 50-70 mL/100 g/min and are postulated to contribute to damage in the penumbra by transiently increasing metabolic demands in this compromised zone. EEG fails at a CBF of below ˜30 mL/100 g/min, hemiparesis is seen at CBF thresholds of ˜23 mL/100 g/min, and anoxic depolarization occurs at CBF values between 15 and 20 mL/100 g/min, resulting in ionic failure. These CBF thresholds for failure of molecular and cellular homeostasis are estimates for the adult brain. Corresponding ischemic threshold values in the developing brain are less well established. Also, threshold values can be a moving target. For example, if neuronal metabolic demands are increased (i.e., hyperthermia, seizures), the CBF




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
