Pharmacogenomic Implication Scenarios
Pharmacotherapy in the ICU is often a complex undertaking with patient-specific variables such as severity of illness, comorbid disease, and volume status greatly altering pharmacokinetic and pharmacodynamic parameters. The latter can often lead to increases in ADRs or alter the effectiveness of many common agents used in ICU, such as analgesics and sedatives, antimicrobials and anticoagulants. Despite the ever increasing number of medications that have pharmacogenomics data and the known variability in response to pharmacotherapy in critically ill patient, scant data is available regarding pharmacogenomics-guided therapy in the ICU. The ICU presents a unique set of challenges with most patients having life-threatening injuries or illness. The high level of acuity necessitates rapid processing of patient information as critically ill patients require constant and close monitoring. Any use of pharmacogenomic-guided pharmacotherapy in the ICU will require rapid turnaround for results, as patients have the potential to deteriorate quickly. It is conceivable that genetic testing, comprising a panel of susceptible genes and their related polymorphisms such as in CYP2D6, could be invaluable in optimizing drug doses in patients; indeed, clinical genotyping has recently become clinically available. However, the efficacy, as well as practical clinical application, particularly in a critical scenario, of this type of testing has yet to be realized.
Anticoagulation Therapy
Warfarin is a vitamin K antagonist widely used to manage thromboembolic disorders such as deep vein thrombosis; prevents strokes and other thromboembolic complications in patients with atrial fibrillation; and prevents venous thromboembolism after major orthopedic surgery. Warfarin has a narrow therapeutic range and a wide variation in dosing requirements; doses can vary up to 10-fold between patients. One of the major ADRs encountered by 10% to 16% of patients is life-threatening bleeding (9). Although initiation of oral anticoagulation is primarily an issue which is dealt with by primary care physicians, since patients in critical care settings are often the most fragile and critical care physicians often are required to manage the bleeding and thrombotic complications of anticoagulants, there is considerable interest to improve time patients spend in the therapeutic range and reduce bleeding complications (10). Dosing adjustments are necessary to maintain therapeutic anticoagulation without triggering bleeding. Adjustments are based on the patient’s prothrombin time (PT), which measures coagulation inhibition. Factors that influence warfarin’s therapeutic and adverse effects include not only nongenetic factors such as gender, age, and body weight, but also significant genetic variants. Warfarin is catabolized by CYP2C9, and exerts its anticoagulant effect partially via inhibition of VKORC1 (vitamin K epoxide hydrolase). CYP2C9*1 is the hepatic enzyme responsible for metabolizing S-warfarin, which is three to five times more potent than R-warfarin (S-warfarin and R-warfarin are enantiomers, or mirror images, of the warfarin molecule). Inactivating polymorphisms in CYP2C9 are common (11), with 2% to 10% of most populations being homozygous for low-activity variants, and are associated with lower warfarin clearance, a higher risk of bleeding complications, and lower dose requirements (12). The two most common variants with reduced enzyme activity among individuals of European ancestry are CYP2C9*2 and CYP2C9*3 which impair metabolism of S-warfarin by about 30% to 40%, and about 80% to 90%, respectively (13). They result in decreased clearance and increased blood levels of S-warfarin (14). Additional CYP2C9 variant alleles with reduced activity (CYP2C9*5, *6, *8, and *11) contribute to dose variability among African Americans. VKORC1 encodes the vitamin K–epoxide reductase protein, the target enzyme of warfarin. VKORC1 catalyzes the conversion of vitamin K–epoxide to vitamin K, which is the rate-limiting step in vitamin K recycling (15). A common noncoding variant (−1639G>A) is significantly associated with warfarin sensitivity and reduced dose requirements, as −1639A carriers require lower initial warfarin doses than −1639G carriers. The −1639G>A polymorphism alters a VKORC1 transcription factor binding site, leading to lower protein expression (16). The −1639G>A allele frequency varies among different ethnic group and largely explains the differences in average dose requirements among whites, blacks, and Asians (10). In 2007, the FDA modified the warfarin label, stating that CYP2C9 and VKORC1 genotypes may be useful in determining the optimal initial dose of warfarin (17,18). The label was further updated in 2010 to include recommendations for initial dosing ranges for patients with different combinations of CYP2C9 and VKORC1 genotypes (Table 154.2). Genetic testing can identify patients who are at greater risk for ADRs so doses can be appropriately adjusted.
| TABLE 154.2 Recommended Daily Warfarin Doses (mg/day) to Achieve a Therapeutic INR Based on CYP2C9 and VKORC1 Genotype Using the FDA-Approved Warfarin Product Insert |
 |
Several novel anticoagulants, including the direct thrombin inhibitor dabigatran and factor Xa inhibitors rivaroxaban and apixaban, have been recently approved. These are being used more widely as they are easier for patients and practitioners since they do not require monitoring of blood levels and frequent dose titration. However, because levels are not monitored, there is concern on the part of many practitioners because significant variability in the levels of active metabolites of some of these medications have been observed (19). Although there is considerable interest in personalizing their application, there is currently no robust data to guide practitioners with respect to novel anticoagulants.
ANTIPLATELET THERAPY
Antiplatelet medications including aspirin and P2Y12 inhibitors, such as clopidogrel, prasugrel and ticagrelor have become essential to management and prevention of a variety of conditions including acute coronary syndrome (ACS), stroke, and peripheral artery disease. Clopidogrel is one of the most commonly used antiplatelet drugs, but up to 30% of patients do not respond adequately to this medication (20). The heterogeneity in response to clopidogrel is largely thought to be due to it being a prodrug with no antiplatelet activity. It requires conversion to an active metabolite with antiplatelet activity by the CYP2C19 enzyme (21). However, certain CYP2C19 variants like the *2 allele which is found in 30% of Asians, 17.3% of blacks, and 14.7% in Caucasians, result in a loss of function (LOF) of the CYP2C19 enzyme. That has been shown to reduce the effectiveness of clopidogrel and is associated with increased rates of stent thrombosis (22,23). The influence of this variant is likely less significant in other clinical scenarios, when clopidogrel is used for indications such as stroke. With the widespread use of clopidogrel in clinical practice and its need to be metabolized to its active form by CYP 2C19, the use of pharmacogenomics to aid in patient selection has been suggested by various organizations. There is concern from both the FDA and the European Medicines Agency (EMA) that a patient’s genotype may significantly influence dosing of many of these medications and therefore added information to the labels of these medications highlighting that genotype may be useful in determining optimal dosing. The Clinical Pharmacogenetics Implementation Consortium (CPIC) recently updated its guidelines on CYP2C19 and clopidogrel to suggest using pharmacogenomics to guide the use of clopidogrel in ACS/PCI patients only (24). The rationale for the CPIC recommendation is the considerable evidence that links CYP2C19 genotype to clinical outcomes in clopidogrel-treated ACS patients; this relationship has yet to be established for non–ACS-related clopidogrel use. The CPIC guidelines also recommend that, based on CYP2C19 genotyping, patients found to be intermediate or poor metabolizers should be discouraged from using clopidogrel and alternative antiplatelet agents such as prasugrel and ticagrelor be used. Patients who exhibit ultra-rapid or extensive metabolism are candidates for standard clopidogrel dosing. The American College of Cardiology/American Heart Association ACS guidelines recommend considering testing for the *2 variant in the setting of recurrent ACS or patients at high risk for poor outcomes (25,26). With the increasing availability and reduced turnaround time of genetic testing for CYP2C19*2 variant, this will likely become used with increasing frequency in the near future. Although other genetic variants of PON1 and CES1 enzymes as well as the ABCB1 multidrug transporter have been associated with poor response to clopidogrel therapy, none have been studied to the extent of CYP2C19*2, have as robust data supporting its significance, or have been included in guidelines for testing in the setting of ACS or stroke (25). The novel P2Y12 inhibitors are less dependent on CPY metabolism, with prasugrel requiring one and ticagrelor requiring no CYP-dependent step for oxidation. This is in contrast to the two-step oxidation required by clopidogrel (25). This may account for the greater benefit seen in patients with CYP2C19 LOF alleles treated with prasugrel over clopidogrel, compared to those without LOF alleles (27). Similarly ticagrelor has been associated with improved outcomes compared to clopidogrel, regardless of CYP2C19 LOF alleles (28). Because this evidence of reduced impact from variable metabolism by CYP450 enzyme family with novel P2Y12 inhibitors, some have argued that these should be used more routinely as their use would preclude the need for genetic testing.
PAIN MANAGEMENT
Genetic factors have been shown to regulate both opioid pharmacokinetics (i.e., metabolizing enzymes and transporters) and pharmacodynamics (i.e., target receptor sites and signal transduction elements) and could be a major contributor to the interpatient variability in response that is observed in clinical practice. The opioids codeine, tramadol, and oxycodone have to be activated by CYP2D6 into their active moieties morphine (29,30), O-desmethyltramadol (M1) (31), and oxymorphone (32), respectively. The CYP2D6 gene is highly polymorphic and displays large interethnic variations (33) with 6% to 10% of Caucasians being completely deficient (34). Individuals with the CYP2D6 poor metabolizer phenotype are unable to metabolize codeine into its more potent morphine metabolite and have little analgesic relief from codeine (35,36). In contrast, individuals who are ultra-metabolizers have gene duplication and may experience potential adverse effects and overdose from the rapid conversion of codeine to its metabolite morphine. Deaths have occurred in children with obstructive sleep apnea who took codeine for pain relief after tonsillectomy and/or adenoidectomy and who had evidence of being ultra-rapid metabolizers of codeine (37). CPIC guidelines for codeine therapy in the context of CYP2D6 phenotype were recently published (38,39) with the recommendation to avoid codeine use in UM and PM for CYP2D6 and consider alternative analgesics such as morphine. Genetic variability in the CYP2D6 gene has been reported to account for some of the interindividual variability associated with methadone (36) and treatment success seems lesser in patients who are ultra-rapid metabolizers than in those who are poor metabolizers. Glucuronidation of morphine into its metabolites is catalyzed by the UDP-glucuronosyltransferase (UGT) enzyme (40). Allelic variations of this enzyme have been associated with variability in hepatic clearance.
More than 100 variants in the μ-opioid receptor gene OPRM1 have been identified, of which 20 have a prevalence greater than 1%. The most common single nucleotide polymorphism of the μ-opioid receptor gene is A118G. It has been associated with greater dose requirements for patients receiving morphine and methadone (41).
Tramadol in its racemic form is metabolized via several pathways, including CYP2D6-mediated oxidation to O-desmethyltramadol, which has a 200-fold greater affinity for μ-opioid receptors as compared with the parent drug (42). The (+)-O-desmethyltramadol is principally responsible for opioid receptor–mediated analgesia, whereas (+)- and (−)-tramadol contribute to analgesia by inhibiting reuptake of the neurotransmitters serotonin and noradrenaline. CYP2D6 poor metabolizers have been shown to have much lower median plasma areas under the concentration–time curve for the active metabolite after a dose of tramadol as compared with extensive metabolizers (31,43). In addition, several prospective clinical trials have shown that, as compared with CYP2D6 extensive metabolizers, poor metabolizers more often fail to exhibit analgesia in response to tramadol. On the basis of this evidence, it is likely that tramadol may have reduced clinical efficacy in CYP2D6 poor metabolizers.
The use of tricyclic antidepressants to treat psychological disorders has declined in part because of the occurrence of undesirable side effects. They are still used to treat depression, but their main therapeutic use is often for pain management (44). Interindividual differences in side effects and treatment response have been associated with variability of tricyclic plasma concentrations (45). Polymorphisms in CYP2D6 and CYP2C19 affect the efficacy and safety of tricyclics, with some drugs being affected by CYP2D6 only, and others by both polymorphic enzymes. Amitriptyline, clomipramine, doxepin, imipramine, and trimipramine are demethylated by CYP2C19 to pharmacologically active metabolites. These drugs and their metabolites, along with desipramine and nortriptyline, undergo hydroxylation by CYP2D6 to less active metabolites. In the case of CYP2D6 and CYP2C19 poor metabolizers, if a tricyclic is warranted, a 50% reduction of recommended starting dose should be considered and therapeutic drug monitoring should be used to guide dose adjustments (46).
ABACAVIR HYPERSENSITIVITY SYNDROME
Signs and symptoms of hypersensitivity to abacavir—an antiretroviral agent—include rash, fever, nausea, vomiting, and dyspnea. The hypersensitivity is strongly associated with the HLA-B*57:0 allele of the human leukocyte antigen B locus (47). The prevalence of the allele is estimated to be 3.4% to 5.8% in populations of European ancestry, 17.6% in Indian Americans, 3.0% in Hispanic Americans, and 1.2% in Chinese Americans. There is significant variability in the prevalence of HLA-B*5701 among African populations. In African Americans, the prevalence is estimated to be 1.0% on average, 0% in the Yoruba from Nigeria, 3.3% in the Luhya from Kenya, and 13.6% in the Masai from Kenya, although the average values are derived from highly variable frequencies within sample groups (48). The FDA strongly recommends genetic testing of HIV-positive patients for the HLA-B*5701 (rs2395029) before they are prescribed abacavir; the screening for this allele substantially reduces the incidence of abacavir hypersensitivity. This drug should be prescribed only for HIV-positive patients who do not have the HLA-B*5701 allele. A hypersensitivity reaction can occur within the first 6 weeks of beginning treatment with abacavir.
IRINOTECAN TOXICITY
Irinotecan is a topoisomerase I inhibitor used to treat solid tumors, especially colorectal cancer in combination with other chemotherapeutic agents. Adverse effects of irinotecan treatment include severe diarrhea, myelosuppression, and neutropenia. These effects are likely induced by inefficient metabolism and excretion of the active metabolite of irinotecan, SN-38, which undergoes glucuronidation primarily in the liver by UGT1A (UDP glucuronosyltransferase 1 family, polypeptide A complex locus) prior to excretion through the kidneys (49). The UGT1A gene is alternatively spliced to produce nine isoenzymes responsible for glucuronidation, which solubilizes compounds for excretion through the kidneys. Genetic variation in UGT1A correlates with adverse events caused by irinotecan toxicity. UGT1A1*28, the most well-characterized variant, is a TA repeat expansion in the promoter of UGT1A1, increasing the number of TA repeats from six to seven; this variant causes reduced levels of UGT1A1 gene expression. UGT1A1*28 occurs at high frequency in white and African populations (26%–31% and 42%–56%, respectively) and at lower frequency in Asians (9%–16%) (50).
Gilbert syndrome is a phenotypic effect often caused by homozygous UGT1A1*28 status (51). It is characterized by mild jaundice due to increased unconjugated bilirubin levels. The role of routine testing for the presence of germline isoforms of UGT1A remains unsettled. At relatively high irinotecan dose levels (>250 mg/m2), patients who are homozygous for the UGT1A1*28 variant experience a greater risk of clinically important neutropenia. However, at the lower doses of irinotecan (100–125 mg/m2), most commonly used in clinical practice, the negative impact of UGT1A1*28 is far more modest and of questionable clinical relevance. It is thus reasonable, but not absolutely mandatory, to determine the UGT1A genetic background to assist in toxicity management for patients scheduled to receive a high-dose irinotecan regimen, or one that combines irinotecan with oxaliplatin.
THIOPURINE TOXICITY
Thiopurine methyltransferase (TPMT) methylates thiopurines such as mercaptopurine, the antileukemic drug that is also the product of azathioprine metabolism. One in 300 individuals is homozygous deficient, 10% are heterozygotes, and about 90% are homozygous for the wild-type TPMT allele (52). Because methylation of mercaptopurine competes with activation of the drug to thioguanine nucleotides, the concentration of the active—but also toxic—thioguanine metabolites is inversely related to TPMT activity and directly related to the probability of pharmacologic effects. Patients with reduced or undetectable TPMT activity treated with standard doses of thiopurines are at risk of serious life-threatening adverse events, including myelosuppression, anemia, bleeding, leukopenia, and severe infection. Dose reductions from the “average” population dose may be required to avoid myelosuppression in 100% of homozygous-deficient patients, 35% of heterozygotes, and only 7% to 8% of those with homozygous wild-type activity (53). The rare homozygous-deficient patients can tolerate 10% or less of the mercaptopurine doses tolerated by the homozygous wild-type patients, with heterozygotes often requiring an intermediate dose. Mercaptopurine has a narrow therapeutic range, and dosing by trial and error can place patients at higher risk of toxicity; thus, prospective adjustment of thiopurine doses based on TPMT genotype has been suggested (54). Life-threatening toxicity has also been reported when thiopurines have been given to patients with nonmalignant conditions (such as Crohn disease, arthritis, or for prevention of solid organ transplant rejection).
ONCOLOGY AS A SPECIAL CASE
Pharmacogenetics in oncology can have a direct severe impact on patient prognosis and treatment depending on whether an individual’s tumor has a specific gene mutation amenable to targeted therapy or not. A good example is non-small cell lung cancer. Approximately 10% of patients with NSCLC in the United States and 35% in East Asia have tumor-associated EGFR mutations (55). These mutations increase the kinase activity of EGFR, leading to hyperactivation of downstream prosurvival signaling pathways. There is a large amount of data supporting sensitivity to EGFR-TKI therapy in patients whose tumors harbor the common exon 19 deletions and L858R point mutations, which make up about 90% of all EGFR mutations (56). Several randomized clinical trials have shown that patients with EGFR mutation-positive tumors have significantly improved progression-free survival and objective response rates when treated with first-line EGFR tyrosine kinase inhibitors (EGFR-TKIs) compared with standard chemotherapy (57). Effective interpretation of EGFR mutation test results thus will help physicians to prescribe the most appropriate treatment for each patient.
SEPSIS
Specific therapies targeting the underlying inflammation causing the circulatory compromise seen in sepsis syndromes, including NSAIDs, tumor necrosis factor (TNF) receptor antagonists, and IL-1 receptor antagonists, have so far been largely unsuccessful in improving mortality (58–60). The failure of these interventions to improve sepsis outcomes may be, in large part, due to the use of anti-inflammatory therapy in a group of patients with very heterogeneous inflammatory responses. The immunologic variability seen in the sepsis syndrome stems not just from the inciting organism and the patient’s comorbidities, but from genetic and epigenetic variation (61). Because the underlying pathophysiology of sepsis is still thought to be driven by immune dysregulation, the heterogeneity of immune responses makes sepsis an ideal candidate for more personalized immunomodulation. Although several genetic and epigenetic profiles have been associated with poor outcomes, due to the acuity of sepsis management, application of these profiles has been limited by turnaround time of the tests (62). However, in recent years the results of these tests are now available within hours as opposed to days or weeks, as was previously the case.
Perhaps the most extensively studied source of immunologic variability in sepsis is SNPs of genes encoding inflammatory modulatory proteins such as TNF and toll-like receptors (TLR). For example, patients who have an adenine for guanine substitution at position –308 in the TNF promoter display increased circulating TNF levels and have decreased risk of acquiring pneumonia (63). However, if infection does occur, the risk of developing septic shock is increased. This finding raises the possibility that treatment with anti-TNF agents in septic patients with –308 mutations could improve outcomes; at this point, this trial has yet to be performed. Although SNPs hold great promise in identifying patients who may benefit from immunomodulatory therapy, there is currently sparse prospective data for their practical application. In addition, SNP studies have been largely retrospective and correlational, therefore they can at best predict, but cannot characterize, the inflammatory milieu at a given time in a patient. This is particularly pertinent as there is increasing evidence that the inflammatory milieu varies greatly between patients, and changes as sepsis progresses, in an unpredictable fashion and does not follow the traditional two-phase model where there is an initial phase of hyperinflammation followed by a hypoinflammatory phase (61). Because defining a patient’s genotype is limited to predicating inflammatory responses of patients and does not necessarily characterize their phenotype, the application of SNP profiles of patients will likely remain limited to predicting a patients’ inflammatory response or risk stratification. Because of this limitation, largely due to epigenetic factors, there has been increasing focus placed on quantifying gene expression and defining inflammatory phenotypes. Perhaps the most promising method to measure genetic expression patterns in the setting of sepsis is mRNA quantification. Quantification of mRNA can be done quickly and provides information regarding expression of a many genes in real time, which is particularly attractive in sepsis where the patient’s condition changes rapidly. Recently these techniques have been applied to pediatric populations and were able to define two distinct expression profiles, one of which was associated with worse outcomes independently of factors such as age, comorbidity, and severity of illness (64). Expression profiles were available with a turnaround time of 8 to 12 hours, making it possible to use this information in near real time. The profile with worse outcomes showed decreased expression of genes involved in adaptive immunity and glucocorticoid receptor signaling. In this group adjunctive corticosteroids were independently associated with poorer outcomes, suggesting that this group of patients either do not respond to corticosteroid administration or outcomes are directly worsened by corticosteroid administration and may account for some of the heterogeneity seen in studies of corticosteroid use in sepsis (65). Although this technique holds promise for selecting patients for steroid therapy, risk stratification, and a number of other applications, it has not been applied in prospective studies.
In addition to using genetic or epigenetic profiles to subclassify patients or select them for therapy, others have measured inflammatory markers directly to both improve diagnostics and identify candidates for specific therapy. Early in the course of sepsis, before a source becomes apparent, it can be difficult to differentiate sepsis from uninfected SIRS. However, the application of procalcitonin levels have been shown to be beneficial in guiding de-escalation of antibiotics in these cases (66). Increased levels of other inflammatory markers, such as IL-6, IL-8, and MCP-1, have also been associated with steroid responsiveness in sepsis, suggesting that these may be used to select patients in which steroids would be of benefit (66,67). Again, these have not been validated in larger prospective studies and thus have not been universally recommended.
Although there are limited applications of personalized medicine in the setting of sepsis at this point, improvements in technology and the resulting improvement turnaround time for a number of genetic and epigenetic tests have allowed for these techniques to be increasingly viable. As these techniques become more widely available, they will almost certainly be applied to sepsis as this is an area where there is great potential for benefit. This, in turn, may usher in a new era where management of patients with sepsis can be individualized to target the underlying pathophysiology, diagnose sepsis more accurately, and modulate the inflammatory response in real time to improve mortality.
GENETICS OF ARDS
Because acute respiratory distress syndrome (ARDS) has a broad definition: acute onset of impaired oxygenation in the setting of bilateral infiltrates without underlying cardiac failure or fluid overload, and can be caused by a variety of insults, development of targeted therapy has been difficult and the mainstay remains supportive care aimed at improving gas exchange (68). Although attempts to delineate inflammatory subtypes of ARDS have had limited success, if they could be established, therapy could target the specific inflammatory mediators driving ARDS in the individual patient and modulate the inflammatory response. Additionally, if genetic risk factors were identified, it may be possible to prevent ARDS or predict poor outcomes and mortality.
Defining inflammatory subclasses of ARDS by measuring inflammatory mediators has been more challenging than for systemic diseases such as sepsis, because inflammation in ARDS is more localized and the inflammatory milieu of the lungs is not fully reflected by the level of inflammatory mediators in peripheral blood (69). Attempts to characterize phenotypes of inflammation by local measurement of inflammatory mediators with either catheter-based direct aspiration or bronchoalveolar lavage (BAL) have also been limited because, among other things, they only provide information regarding the alveolar compartment. Measurements of inflammatory markers using BAL including TNF-α, IL-1β, IL-6, IL-8, GRO-α, and MCP-1 have not shown any of these to predict outcomes in ARDS (70). Although measuring inflammatory mediators directly has thus far been unsuccessful at identifying targets for specific therapy or predicting outcomes, there are almost certainly inflammatory phenotypes associated with ARDS that require other methodology to define.
Our ability to define genetic risk factors has greatly increased in recent years. The first gene described as influencing ARDS susceptibility was a nonsynonymous coding variant in surfactant protein B (SFTPB) (71). As this mutation is relatively uncommon, any benefit seen in these patients in large trials of exogenous surfactant administration would likely not have been great enough to influence the overall outcomes of the patients who received surfactant therapy. However since these patients have dysfunctional surfactant production, they would theoretically benefit from supplementation with exogenous surfactant while individuals with normal surfactant production may not. Some risk factors also appear to be only, or more, significant in the setting of specific inciting circumstances. For example, individuals with IL-8 promoter variant (rs4073A), which results in persistent IL-8 elevations, had an increased ARDS incidence after major trauma (72). However, this association has not been seen in ARDS due to sepsis, suggesting that different inflammatory pathways are involved in the pathogenesis of ARDS. Despite the heterogeneity, there does appear to be some overlap in inflammatory response. This was seen in an SNP in the IL-1 receptor antagonist (IL-1RA) gene (rs315952) which is associated with lower risk of ARDS in patients with either severe trauma or septic shock (73). This SNP was associated with higher levels of IL-1RA, therefore it is possible that some patients, especially those without this mutation, could benefit from treatment with IL-1RA either for therapeutic or prophylactic purposes.
While some polymorphisms are associated with poor outcomes across different ethnicities, other risk factors appear to be associated with certain genetic populations. The ARDS Network has demonstrated that the absolute risk of mortality was increased by 17% in those patients who do not express Duffy antigen (Duffy null), compared to those with at least one wild-type Duffy gene (74). This mutation is much more common in patients of African descent, with 60% to 80% being Duffy null, as it confers resistance to the malaria-causing organism Plasmodium vivax (71). The increase in mortality seen in those with this mutation is believed to be a result of higher levels of inflammatory cytokines, such as IL-8, as a result of the absence of Duffy antigen, which normally functions as a chemokine scavenger.
PHARMACOGENOMIC TESTING
Two approaches are now available to help personalize drug therapies. Phenotyping and genotyping tests are used in clinical practice to identify variations in CYP enzymatic activities or CYP allelic variants. They may help in predicting the right dose for the right patient, and anticipating toxicities or therapeutic inefficacies. Phenotyping consists of the administration of a probe drug metabolized by a specific CYP enzyme. The assessment of different pharmacokinetic parameters of the probe drug and its metabolites or the determination of a ratio between the drug and its metabolite (metabolic ratio) allow the definition of an individual metabolic profile.
Individual phenotyping involves the administration of one CYP-specific probe, whereas during simultaneous phenotyping, a mixture of probes is administered concomitantly which allows the concurrent detection of the activity of multiple enzymes. Several drugs (debrisoquine, sparteine, dextromethorphan) have been employed as CYP2D6 phenotyping probes (75,76). CYP phenotyping provides information on the real-time (in vivo) activity of CYP enzymes and may therefore provide the most clinically relevant information as it reflects a combination of genetic, environmental, and endogenous factors.
Genotyping allows precise determination of the individual DNA sequence and analysis of functional genetic mutations coding for specific enzymes. It offers the possibility of predicting the phenotype based on the alleles identified provided that the relationship between genotype and phenotype has been established. Its major drawback is the incapacity to measure the influence of the environmental factors, such as drug–drug interactions, on the enzymatic activity. Different molecular biologic methods can be utilized to analyze DNA sequence coding for genes relevant in drug metabolism. Single analyte assays usually target just one gene covering a small subset of variant alleles. Real-time qPCR-based assay methods for most clinically important variant genes of CYP2C9, CYP2C19, and CYP2D6 are now available. Depending on the number of SNPs to be screened, multiplexing approaches have also been developed. In contrast, a multianalyte assay like microarray technologies (Roche AmpliChip CYP450 GeneChip®) or next-generation sequencing offer the possibility to analyze higher number of genes in one run. An important variable in the use of these assays is turnaround time (TAT). Single analyte assays based on qPCR can be easily turned around during the same day, but microarrays and sequencing are hampered by elaborate and time-consuming (several-day) workflows that prevent them from delivering timely results. The specific method used in a study or setting usually depends on the requirements of the study, ranging from determining one or two SNPs in 1 to 10 patients, one or two SNPs in thousands of patients, several thousands of SNPs in an individual patient, or thousands of SNPs in thousands of patients. A very popular category of single analyte assays is based on qPCR; the advantage of these tests is that the target is detected during DNA amplification eliminating a separate detection step. The qPCR instruments are widely available, and laboratory-developed assays can be designed and run easily, if qualified high laboratory personnel are available. Other frequently used tests have two steps: PCR amplification of the target sequence and then detection of the amplified target by various techniques. The detection techniques can be microsphere-based array (Luminex) (77), hybridization capture array (Autogenomics), or DNA hybridization and electrochemical detection (GenMark) to name a few. Some of the new instruments are much simpler to run, fast, and do not require a separate DNA extraction step (Spartan RX CYP2C19).
INHERITED DISORDERS OF METABOLISM
Inherited disorders of metabolism comprise a heterogeneous group of genetic diseases, several of which present in acute metabolic distress, which require emergency management and critical care monitoring. Each disorder involves a defective enzyme or transport protein that normally contributes to proper biochemical process within the body. As a group, the clinical manifestations of metabolic disorders can be extremely variable depending on the affected biochemical pathway, severity of the molecular defect (e.g., the amount of residual enzyme activity), and environmental factors (e.g., illness, fasting, or dietary intake). Although there are approximately 1,000 metabolic disorders described, each disorder is relatively rare; collectively, however, there is an estimated incidence of about 1:4,000.
Increased awareness and detection in all ages, as well as advances in our understanding and management of metabolic diseases, has decreased morbidity and increased survival in children with metabolic disorders, even into adulthood (78,79). In addition to improved early detection of presymptomatic infants with metabolic disorders, newborn screening has secondarily identified affected siblings and family members of all ages. Furthermore, case reports of adult-onset metabolic disease are not uncommon, typically described in the setting of an acute metabolic stressor such as a prolonged presurgical fast or an illness with vomiting and dehydration (80,81). It is essential for the intensivist to be knowledgeable about the possible presentation, initial evaluation, and emergency management for patients of all ages with this group of disorders.
Metabolism refers to the sum of the process of biochemical synthesis (anabolism) and breakdown (catabolism) of compounds, such as proteins, carbohydrates, and lipids from the diet or stored within the body. Inherited metabolic disorders are genetic defects that affect an enzyme or a transport protein important to normal metabolism. Clinically, inherited metabolic disorders manifest with a wide range of symptoms, from chronic progressive disease to acute metabolic crises, following an apparently asymptomatic interval. Depending on the severity and type of disorder, any number of organ systems may be involved. Hepatic failure may present in tyrosinemia type I, Wilson disease, and Gaucher disease. Cardiomyopathy may be a feature in infantile and juvenile Pompe disease, very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency, and various lysosomal storage diseases. Muscle disease, either myopathy and/or rhabdomyolysis, may occur in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), VLCAD deficiency, and McArdle disease. Acute encephalopathy is a concern in many organic acidurias, urea cycle defects, and fatty acid oxidation defects.
Often, the acute presentation of an inherited metabolic disorder may mimic a more common, systemic disease such as sepsis or intoxication; however, swift recognition and management of a metabolic disorder is important for improving the overall morbidity and mortality. Saudubray et al. (82) loosely classified these disorders into three groups.
- Metabolic disorders of intermediary metabolism that cause toxic accumulation of metabolites. This may be conceptualized as a roadblock in the normal metabolic pathway causing a traffic jam and the buildup of potentially toxic intermediary metabolites. Some of these metabolites will be directed to alternative pathways, which may lead to the accumulation of toxic by-products (Fig. 154.1). Examples of this group include organic acidurias, aminoacidopathies, fatty acid oxidation defects, urea cycle defects, disorders of metal transport (e.g., Wilson disease), and carbohydrate defects. In critical care, many of these diseases present in acute metabolic crises with some combination of encephalopathy, liver disease, multisystem failure, metabolic acidosis, ketoacidosis, lactic acidosis, or hyperammonemia.

FIGURE 154.1 Most inborn errors of metabolism are due to an enzyme defect within a metabolic pathway. The defect may lead to toxic accumulation of substrate, or metabolic by-products, or to deficiency of an essential product within that pathway.
- Metabolic defects that affect cellular respiration or mitochondrial energy production. This group includes enzyme defects of the mitochondrial respiratory chain itself. It also includes enzymes in glycolysis, glycogenosis (utilization of glycogen stores for energy), gluconeogenesis, and the tricarboxylic acid (TCA or Kreb) cycles. Fatty acid oxidation disorders, which cause toxic accumulation of fatty acids and other by-products (Group 1), are also considered here, as the products of these reactions are an essential energy source for the TCA cycle and mitochondrial respiratory chain, particularly during fasting stress.
- Disorders involving the synthesis or breakdown of complex (large) molecules. This group includes lysosomal storage disorders, peroxisomal disorders (e.g., X-linked adrenoleukodystrophy), disorders of glycosylation, and cholesterol synthesis defects. In general, these disorders are chronic progressive diseases, and though they may rapidly worsen during illness or stress, they rarely present in acute metabolic crises.
For an exhaustive discussion of all known metabolic disorders, the reader is referred to comprehensive reviews (83,84). Herein, we focus on metabolic disorders and associated scenarios that may present to the critical care unit.
FATTY ACID OXIDATION DISORDERS
Fatty acid oxidation disorders encompass a group of metabolic defects of mitochondrial β-oxidation. β-Oxidation is the process of breaking down fatty acids to aid in production of adenosine triphosphate (ATP) via the respiratory chain complex. Several two-carbon molecules (acetyl-CoA) are also produced by β-oxidation of each fatty acid. Acetyl-CoA can be used in the tricarboxylic acid cycle for aerobic respiration or as a precursor for the production of ketone bodies. Therefore, the β-oxidation cycle is essential for the normal physiologic response to fasting after typical energy sources, such as glucose and glycogen, are depleted. In the fasting state, vital organs, in particular the brain, require an alternate source of energy in the form of ketone bodies.
Several enzymes are essential in the β-oxidation of fatty acids. Carnitine palmitoyltransferase 1 (CPT1), carnitine palmitoyltransferase 2 (CPT2), and carnitine acylcarnitine carrier protein are important for the transfer of fatty acids into the mitochondria. Long-chain fatty acids (12–18 carbon-length molecules) are acted on by the enzyme’s VLCAD, mitochondrial trifunctional protein, and long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD). Medium-chain fatty acids (6–12 carbon-length molecules) are broken down by the enzyme medium-chain acyl-CoA dehydrogenase (MCAD) (Fig. 154.2).
Disease manifestation, age of onset, and severity vary greatly, depending on the enzyme defect. The most common of these conditions, MCAD deficiency, with an estimated incidence of 1 in 15,000, can present at any age, with fasting intolerance, metabolic encephalopathy, and hypoketotic hypoglycemia. Often, the acute presentation is a result of an illness with vomiting and dehydration. This condition may also cause liver disease with significant hyperammonemia, referred to as Reye-like syndrome. Of note, myopathy and cardiomyopathy are rare in MCAD deficiency. Individuals with MCAD deficiency are essentially completely asymptomatic between acute episodes. Long-chain fatty acid defects (VLCAD, LCHAD, or TFP [trifunctional protein] deficiency) and CPT2 deficiency may also present early in childhood with similar fasting intolerance and hypoketotic hypoglycemia, along with cardiomyopathy. However, milder forms of these diseases may not present until adolescence or early adulthood, typically with only muscle disease associated with rhabdomyolysis and/or cardiomyopathy.

FIGURE 154.2 Fatty acid oxidation occurs primarily in the mitochondria. Carnitine plays an important role in transport of long-chain fatty acids into the mitochondria. Each fatty acid molecule is broken down by several enzymatic cycles (β-oxidation) in which each cycle produces a two-carbon molecule (acetyl-CoA). β-Oxidation enzymes are fatty acid chain length specific (i.e., medium-chain acyl-CoA dehydrogenase breaks down fatty acids that are of medium chain length, 6–10 carbons). Acetyl-CoA is important for ketogenesis, energy production, and other essential biochemical roles.
ORGANIC ACIDURIAS AND AMINOACIDOPATHIES
Organic acidurias are caused by defects in the normal cellular breakdown of amino acids or odd-chain fatty acids. Examples include propionic aciduria and methylmalonic aciduria (MMA), which are disorders of the breakdown of the amino acids isoleucine and valine. Other disorders include isovaleric aciduria, 3-methylglutaconic aciduria, glutaric aciduria type I, multiple carboxylase deficiency, and biotinidase deficiency. Aminoacidopathies, disorders marked by the abnormal accumulation of specific amino acids, can be included within this group, as there is clinical overlap in many of these conditions. Amino acids are organic acids that are unique in that they have an amino group. As a group, aminoacidopathies can have much more variability in clinical presentation. Examples may include maple syrup urine disease (MSUD), a defect involving the catabolism of the branched chain amino acids leucine, isoleucine, and valine, or tyrosinemia type I, which can cause accumulation of the by-product succinylacetone, a metabolite toxic to liver cells.
In the ICU, it is important to be aware of this group of disorders, many of which present with acute metabolic crises that may mimic or accompany more typical scenarios such as sepsis or drug overdose.
UREA CYCLE DISORDERS
Defects of the urea cycle affect the normal detoxification of ammonia (Fig. 154.3). Ammonia primarily results from nitrogen waste accumulated from excess intake of protein or the endogenous catabolism of protein. The most common urea cycle disorder, X-linked ornithine transcarbamylase (OTC) deficiency, severely affects neonatal males shortly after birth. This often presents as significant encephalopathy and respiratory alkalosis at a few days of life, after significant protein intake. Females who are carriers for OTC deficiency are essentially heterozygous for the condition but may also be symptomatic, with significant recurrent episodes of hyperammonemia, depending on the extent of X-inactivation. It is not uncommon for females with OTC deficiency to present in later childhood or as adults. Additional urea cycle disorders include citrullinemia, carbamylphospate synthase-1 deficiency, and argininosuccinic aciduria. Acute hyperammonemia in urea cycle defects may manifest at any age. Thus, an individual with poorly explained encephalopathy, including lethargy or coma, should have an ammonia level evaluated.
MITOCHONDRIAL DISEASE
Mitochondrial disorders consist of disorders of oxidative phosphorylation, including pyruvate dehydrogenase deficiency, disorders of the tricarboxylic acid cycle, and respiratory chain defects. These are a heterogeneous group of metabolic defects and should be considered whenever unrelated organ systems are affected without a reasonable explanation. Mitochondrial disorders can be caused by gene defects within the nuclear DNA, which have a typical Mendelian inheritance pattern. In general, these conditions are inherited autosomal recessive and have a clinically more severe onset, usually in infancy and early childhood. Mitochondrial disorders that are inherited from genes within mitochondrial DNA itself are more commonly progressive and present later in life (85).
Defects involving mitochondrial DNA are only maternally inherited, because only the egg—not the sperm—donates mitochondria to the zygote. Another consideration in mitochondrial (maternal) inherited disorders is that each cell in the body contains multiple sets of mitochondria; therefore, the same cell—and thus an individual—may have a mixture of both mutated and normal mitochondrial DNA. This is referred to as heteroplasmy and contributes to the phenotypic heterogeneity observed in patients with maternally inherited mitochondrial disorders. The clinician should be aware of mitochondrial disorders, such as the more common MELAS, which may present to the ICU with multisystem disease. Of particular concern in these disorders is metabolic stroke and lactic acidosis (see below).
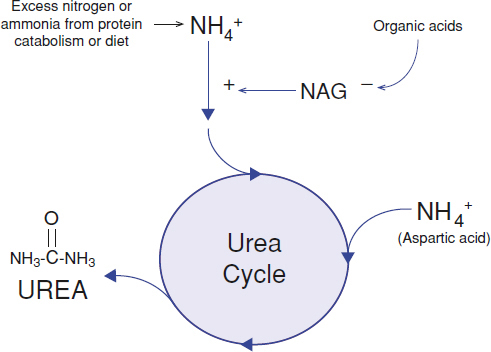
FIGURE 154.3 Removal of excess nitrogen (ammonium, NH4+) waste from dietary intake and catabolism of protein is facilitated by several enzyme steps in the urea cycle. The end product is a water-soluble compound, urea, which is readily excreted by the kidneys. N-Acetylglutamate (NAG) is an important positive activator of carbamoyl phosphate synthetase, the first enzyme in the urea cycle. The formation of NAG is inhibited by accumulation of organic acid metabolites from the breakdown of organic acids, amino acids, fatty acids, and other organic compounds, thus potentially leading to secondary hyperammonemia.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


