 There are many causes of metabolic encephalopathy, so that a systematic investigation, including laboratory testing and neuroimaging, is essential.
There are many causes of metabolic encephalopathy, so that a systematic investigation, including laboratory testing and neuroimaging, is essential. The practitioner should not confuse a symptom or a syndrome (such as Reye-like illness) with an etiology.
The practitioner should not confuse a symptom or a syndrome (such as Reye-like illness) with an etiology. Diseases that are amenable to treatment should be considered first.
Diseases that are amenable to treatment should be considered first. Intracranial hypertension is frequent in metabolic encephalopathy and requires strict attention to whether hemodynamic support is needed.
Intracranial hypertension is frequent in metabolic encephalopathy and requires strict attention to whether hemodynamic support is needed. Specific treatment of endogenous intoxication must be started as soon as possible and conducted simultaneously with diagnostic investigations.
Specific treatment of endogenous intoxication must be started as soon as possible and conducted simultaneously with diagnostic investigations.neurons. The downregulation of SNAT5 can result in a decrease in excitatory neurotransmission in the brain and lead to excessive neuroinhibition, which is characteristic of encephalopathy in hyperammonemia (5,6,7,8). Glutamine is also thought to disrupt mitochondrial permeability by interference with the mitochondria-permeability-transition process and liberation of free radicals through hydrolysis of ammonia, all subsequently leading to mitochondrial swelling and dysfunction (9). Another hypothesis for hyperammonemia-induced encephalopathy is injury through the effect of increased production of glutamine that leads to an excess of extracellular glutamate, which induces neuronal hyperexcitability through activation of a subgroup of the glutamate family of receptors, the N-methyl-D-aspartate (NMDA) ionotropic receptors. Activation of NMDA receptors leads to depolarization and downstream changes in nitric oxide (NO) metabolism, disturbances in sodium/potassium adenosine triphosphatase (Na+/K+-ATPase) function, mitochondrial dysfunction, energy failure, and oxidative stress, together causing cell death (10). Finally, a high level of ammonia has the potential for participating in causing brain injury and cerebral edema by inducing hyperemia in cerebral blood flow (CBF) and by impairing cerebral autoregulation (11,12).
TABLE 66.1 CAUSES OF METABOLIC ENCEPHALOPATHIES CLASSIFIED ACCORDING TO PATHOPHYSIOLOGY | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
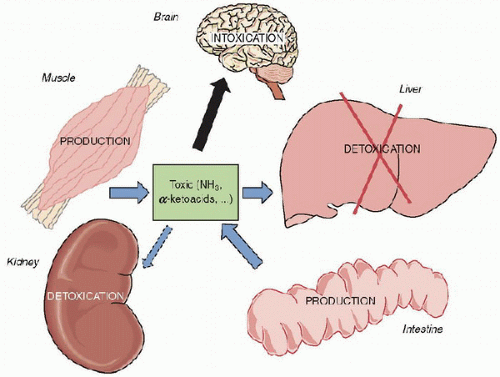 FIGURE 66.1. Endogenous intoxication model: impairment of nitrogen metabolism due to liver failure or inborn error of catabolic pathway of amino acids. The nitrogen produced by the intestine and muscle amino acid catabolism is not properly metabolized, resulting in toxic accumulation and brain damage. (Adapted from Rabier D [Biochemical Laboratory, Necker Hospital, France].) |
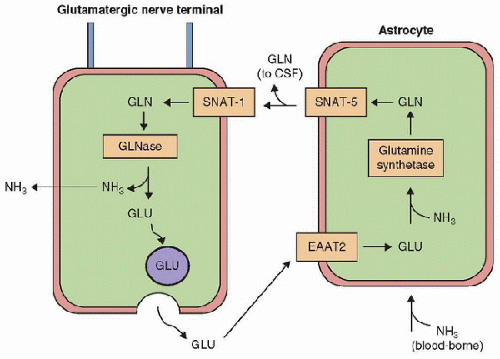 FIGURE 66.2. Schematic representation of a glutamate/glutamine cycle. The figure illustrates position of the astrocytic glutamine synthetase (GS), neuronal glutaminase (GLNase), the astrocytic glutamate transporter EAAT-2, and the astrocyte and neuron. (From Desjardins P, Du T, Jiang W, et al. Pathogenesis of hepatic encephalopathy and brain edema in acute liver failure: Role of glutamine redefined. Neurochem Int 2012;60(7):690-6, with permission.) Gln, glutamine; Glu, glutamate; NH3, ammonia; CSF, cerebral spinal fluid. |
 and clinical expertise to identify other potential conditions. Therefore, whether Reye syndrome should be accepted as a diagnostic label before all other possible causes have been excluded is debatable (14). In this setting, we prefer to use the term “Reye-like illness.”
and clinical expertise to identify other potential conditions. Therefore, whether Reye syndrome should be accepted as a diagnostic label before all other possible causes have been excluded is debatable (14). In this setting, we prefer to use the term “Reye-like illness.”ligand levels in patients with hepatic encephalopathy, was first considered a possibility when similarities between evoked potential in rats with liver failure and animals treated with GABA receptor agonists were noted (13). Animal studies in models of hepatic encephalopathy have also demonstrated a beneficial effect on arousal of administering flumazenil, a GABA receptor antagonist. A few studies in humans have shown the same benefit (20), but the origin of increased GABA receptor agonists in hepatic encephalopathy is unclear; it is possible that the agonist or a precursor is contained within the food cycle, is synthesized by gastrointestinal flora, or that there is occult ingestion of pharmaceutical benzodiazepine (21,22). It is also possible that endogenous levels of the neurosteroid hormone dehydroepiandrosterone sulfate, found to be low in humans with cirrhosis, may contribute to increased GABAergic tone. Improvement in encephalopathy after administering flumazenil supports this hypothesis. However, the improvement is transient and incomplete, which demonstrates the importance of other toxic mechanisms in the pathogenesis of hepatic encephalopathy, such as glutamine-induced astrocytic swelling (see above).
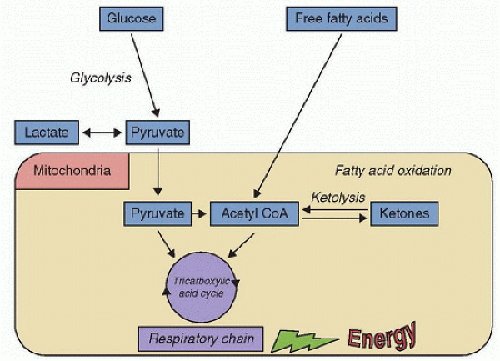 FIGURE 66.3. Energy failure model of metabolic encephalopathy. Hypoglycemia and any enzyme defect in mitochondrial energy production may result in metabolic encephalopathy. The pathophysiology of metabolic encephalopathy by fatty acid oxidation defect is multifactorial. |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


