1. Hypoglycemia is a common concern. Preoperative fasting must be minimized, and intravenous dextrose must be supplemented in the perioperative period.
2. Lactated Ringer solution should not be used in patients with organic acidemias or patients at risk for lactic acidosis with inappropriate lactate metabolism.
3. Arterial lines should be considered for disorders requiring frequent evaluation of electrolyte or acid–base status.
4. The incidence of pulmonary aspiration is frequently higher in these patients than in the general population.
5. Hepatomegaly (with hepatic dysfunction) and hypersplenism may result in coagulopathy resulting from thrombocytopenia, reduced clotting factor synthesis, or increased fibrinolysis. Liver transplantation may be curative for some organic acidemias and urea cycle disorders.
6. Seizure disorders are common among patients with inborn errors of metabolism, and anticonvulsants should be continued perioperatively. The metabolism of anesthetic drugs and skeletal muscle relaxants may be affected by chronic anticonvulsant use.
7. Sevoflurane can produce epileptiform activity during the induction of anesthesia. The safety of an inhaled anesthetic induction with sevoflurane in patients with seizure disorders remains unclear; however, most pediatric anesthesiologists continue to use sevoflurane in these patients without incident.
Plans for the anesthetic management of patients with metabolic diseases and inborn errors of metabolism must consider the unique pathophysiology of each disorder; yet ironically, many of the anesthetic implications for these disorders will overlap.
A clinician will not encounter many of these disorders during his or her entire career. Accordingly, many text and Internet references are available for the anesthesiologist to obtain additional detailed information. The reference texts Anesthesia for Genetic, Metabolic, and Dysmorphic Syndromes of Childhood by Baum and O’Flaherty and Syndromes: Rapid Recognition and Perioperative Implications by Bissonnette et al. are excellent resources for the practitioner faced with an unfamiliar error of metabolism (1,2). The online database Online Mendelian Inheritance in Man (OMIM), hosted by the National Library of Medicine at the National Institutes of Health, provides a comprehensive review of genetic, diagnostic, and clinical management features of inherited disorders, and is available at http://www.ncbi.nlm.nih.gov/omim. Lastly, the online expert-authored textbook GeneReviews® (3) provides an easily accessible and comprehensive review of the characteristics, diagnosis, and management of many inherited conditions. It is available for free at http://www.ncbi.nlm.nih.gov/books/NBK1116/.
This chapter is not intended to be comprehensive, but rather, is a review of the clinically applicable anesthetic implications of the most frequently encountered metabolic diseases and inborn errors of metabolism. The care of these complex and unusual patients should be coordinated with a specialist in metabolic diseases, and a comprehensive preoperative, intraoperative, and postoperative plan should be formulated with the entire team.
DISORDER: Amino acid metabolism
NONBRANCHED CHAIN
1. Cystinosis is an intracellular accumulation disorder of cystine that typically leads to systemic hypertension, renal tubular Fanconi syndrome, and chronic renal insufficiency. Medical management with cysteine-depleting therapy (cysteamine) ameliorates or delays systemic complications (3).
a. Hepatomegaly is common, but cirrhosis and portal hypertension rarely result from accumulation in the liver (4). Hypothyroidism and diabetes mellitus are also common.
b. Perioperative electrolyte levels (including potassium, bicarbonate, calcium, and glucose) must be monitored closely.
c. Isosthenuria and polyuria may complicate intravascular volume assessment; therefore, cases with large fluid shifts merit central venous pressure monitoring.
d. Gastric acid hypersecretion is common with cysteamine therapy (3), and the risk of pulmonary aspiration is likely elevated.
e. Depolarizing muscle relaxants should be avoided to prevent succinylcholine-induced hyperkalemia in patients with associated hypotonia and myopathy.
f. Some of the patients are photophobic; therefore, it may be beneficial to dim the operating room lights until after induction (2).
2. Cystinuria, or diamino pentosuria, is a disorder of increased renal clearance of certain amino acids, including cysteine.
a. Nephrolithiasis and chronic renal insufficiency are associated with the disease.
b. Serum potassium, bicarbonate, and volume status should be evaluated perioperatively.
c. Perioperative diuresis and chronic urine alkalinization should be continued (5).
3. Homocystinuria is a triad of diseases involving methionine metabolism that leads to disordered collagen production and hypercoagulability (6).
a. Common features include mental retardation, marfanoid habitus, high-arched palate, ectopia lentis, and pectus excavatum.
b. Spontaneous thromboemboli may be venous or arterial and commonly affect the coronary, cerebral, renal, mesenteric, and pulmonary vasculature (6).
c. Prolonged fasting may exacerbate hypercoagulability and hyperinsulinemia-induced hypoglycemia; therefore, dextrose-containing intravenous fluids should be initiated preoperatively and continued at 1.5 times maintenance (3).
d. Perioperative deep venous thrombosis risk is elevated and heparin appears to be ineffective. Pyridoxine and dipyridamole may be superior pharmacologic prophylaxis (1).
e. Nitrous oxide is relatively contraindicated, as inhibition of methionine synthase increases homocysteine levels and may induce cerebral atrophy, central nervous system demyelination, and arterial thrombosis (7–9).
f. Seizures are present in 21% of affected individuals. Psychiatric disorders are common. Anticonvulsants should be continued perioperatively (3).
CLINICAL PEARL Prolonged fasting may exacerbate hypercoagulability and hyperinsulinemia-induced hypoglycemia; therefore, dextrose-containing intravenous fluids should be initiated preoperatively and continued at 1.5 times maintenance (3).
4. Phenylketonuria is a disorder of phenylalanine metabolism.
a. Common manifestations of untreated disease include seizures and psychomotor retardation.
b. Anticonvulsant medications should be continued in the perioperative period.
c. Strict dietary avoidance of phenylalanine from infancy until at least early adulthood will moderate the disease process (1).
d. Dietary protein restrictions may lead to vitamin B12 deficiency, and nitrous oxide should be avoided to prevent myeloneuropathy in these patients. Serum B12 measurements are insensitive, so hemoglobin, methylmalonate, and homocysteine levels should be evaluated (10,11).
e. Fasting and stress should be minimized to prevent lean muscle catabolism and liberation of phenylalanine (1).
5. Tyrosinemia I is characterized by abnormal tyrosine metabolism, and frequently presents with renal tubular dysfunction and hepatic failure or cirrhosis.
a. Medical management includes the enzyme inhibitor nitisinone that diverts tyrosine metabolism to alternate pathways. Side effects include thrombocytopenia and leucopenia and a preoperative cell blood count is indicated (12).
b. Fasting hypoglycemia is a concern, and dextrose-containing intravenous fluids should be administered. Hypoglycemia may be unresponsive to glucagon (1).
c. Renal tubular dysfunction mandates careful fluid management and evaluation of perioperative hemoglobin, bicarbonate, and potassium levels.
d. Coagulopathy may be profound and unresponsive to vitamin K.
e. Polyneuropathy and extensor hypertonia are common, and infection may precipitate profound neuromuscular weakness requiring mechanical ventilation.
f. Hypertrophic cardiomyopathy and systemic hypertension have been described.
g. Symptomatic “porphyria-like” crises and toxic metabolite inhibition of porphobilinogen synthesis suggest precautions similar to porphyria be considered (1).
6. Alcaptonuria is a disorder of tyrosine and phenylalanine metabolism that results in bluish-black pigmentation in the connective tissues (ochronosis).
a. Aortic and mitral valve calcification and stenosis are possible. Premature coronary artery calcifications should be suspected (3).
b. Spine and large joint arthritis is common and occurs at younger ages (1).
c. Oxidation of metabolites in the urine cause dark discoloration with alkalinization or upon standing.
d. About 50% of patients are affected by nephrolithiasis (3).
7. Hyperoxaluria, or oxalosis, is a disorder of oxalate crystal production and tissue deposition involving the faulty peroxisomal conversion of glyoxylate to glycine (3).
a. Intravascular deposition leads to coronary and peripheral ischemia, and intracardiac deposition may lead to conduction abnormalities or heart block.
b. A preoperative electrocardiogram is recommended.
c. Nephrolithiasis is universal, bone pain and fractures are frequent and systemic hypertension is common (3).
d. Renal tubular acidosis and renal failure are common in this disorder, and careful fluid and electrolyte management is critical. Dialysis is not effective at removing oxalate (1).
e. Adequate hydration, urine alkalinization, and diuretics are essential (in the absence of end-stage renal disease).
f. Preoperative serum bicarbonate, calcium, and potassium should be evaluated.
g. Arterial lines may be beneficial for hemodynamic and electrolyte monitoring, but pose an increased ischemic risk in the presence of peripheral vasculopathy.
h. Pyridoxine is therapeutic in up to 30% of patients (13).
8. Branched-chain organic acidemias
As a group, the branched-chain organic acidemias share many of the same perioperative implications.
a. All disorders involve a significant risk of perioperative metabolic acidosis. Patients will likely be on protein-restrictive diets and may be taking chronic oral citrate or bicarbonate (1).
b. Prolonged fasting should be avoided, and intravenous solutions should contain dextrose to minimize protein catabolism and bicarbonate to replace the daily therapeutic dose.
c. Lactated intravenous solutions may exacerbate the metabolic acidosis and should not be used.
d. The addition of L-carnitine to intravenous fluids may help to further minimize protein catabolism and may improve metabolite excretion by conjugation.
e. Arterial lines are recommended for longer procedures to permit frequent monitoring of acid–base, lactate, and glucose levels.
f. During surgeries in the oropharynx or nasopharynx, where blood may be swallowed, throat packs and orogastric suctioning are important to minimize gastrointestinal blood absorption and to decrease the protein burden.
g. Patients with organic acidemia have an increased risk of pancreatitis (1).
h. Isovaleric acidemia is due to a defect in leucine metabolism, and the elevated isovaleric acid levels produce a distinctive odor similar to “sweaty feet” (3).
(1) Common associated conditions include mental retardation and hypocalcemia.
(2) Monitoring of serum calcium is essential in the perioperative period, especially once the acidemia is corrected.
(3) Pancytopenia and thrombocytopenia may be associated, and a preoperative cell blood count should be considered (1).
(4) Acetylsalicylic acid may worsen this disorder and is contraindicated.
(5) Patients may have associated carnitine deficiency, which may increase their risk for bupivacaine-induced cardiotoxicity; carnitine supplementation should be considered (14).
i. Maple syrup urine disease is a disorder of branched-chain amino acid metabolism that leads to severe metabolic acidosis and hypoglycemia.
(1) Cerebral edema can occur, and can be exacerbated by overly aggressive hydration.
(2) Lipid infusions to maintain caloric intake during prolonged fasting should be considered (15).
(3) Neurologic decline can be acutely precipitated by stress (16).
j. Propionic acidemia is a disorder of amino acid and fatty acid metabolism caused by mitochondrial propionyl coenzyme carboxylase deficiency that leads to hyperammonemia, metabolic acidosis, developmental delay, and hypotonia.
(1) Cardiomyopathy and arrhythmias (including prolonged QTc) may be associated (3).
(2) Gastroesophageal reflux, vomiting, and an impaired gag reflex predispose these patients to pulmonary aspiration and prolonged ventilation.
(3) Seizures are frequent and anticonvulsants should be continued.
(4) Muscle relaxants that are metabolized by ester hydrolysis may precipitate ketoacidosis and should be avoided (17).
(5) There are no data at this time regarding remifentanil, which is metabolized by nonspecific esterases. Case reports suggest that it may be safe (18).
(6) Propofol use is relatively contraindicated, as the emulsion contains large amounts of polyunsaturated fats that are partially metabolized to propionic acid (19).
(7) Propionic acid derivatives (such as ketorolac, ibuprofen, naproxen, and ketoprofen) should be avoided (18).
(8) Neutropenia is common and should be assessed (3).
(9) Oral antibiotics may be utilized to reduce propionate production by gut bacteria (3).
k. Methylmalonic acidemia is a metabolic enzymopathy that is functionally “downstream” from propionic acidemia that presents as potentially severe metabolic acidosis, hypotonia, and osteoporosis. Clinical presentation includes periods of relative health with sporadic decompensation with illness or stress (3).
(1) Associated anomalies include pathologic fractures and pancytopenia in 50% of patients. Accordingly, careful positioning is vital and preoperative blood cell counts should be evaluated.
(2) Similar to propionic acidemia, propofol and drugs metabolized by ester hydrolysis are contraindicated. In addition, oral antibiotics may be utilized to reduce propionate production by gut bacteria (3).
(3) One form is vitamin B12-responsive and nitrous oxide should be avoided (3).
(4) Patients have an increased risk of chronic renal insufficiency.
(5) Hyperkalemia may be caused by acute metabolic decompensation, even with normal renal function (20).
(6) Preoperative hemodialysis may be necessary to correct profound hyperammonemia or acidosis (21).
(7) Cardiomyopathy, hepatomegaly, and growth retardation have been associated with this disorder (1).
l. α-Ketoglutarate dehydrogenase deficiency is a disorder characterized by severe hypotonia and metabolic acidosis.
(1) Evaluation of serum bicarbonate, potassium, glucose, and calcium should be performed preoperatively.
(2) Intravenous hydration is important, but lactated solutions must be avoided.
(3) Prolonged neuromuscular block is possible with nondepolarizing relaxants, and succinylcholine should be avoided (2).
m. Glutaric aciduria I is a disorder of organic acid excretion that leads to chronic neurologic deterioration.
(1) Symptoms include dystonia, macrocephaly/hydrocephalus, and seizures.
(2) Bulbar symptoms, which include swallowing dysfunction, inability to manage oral secretions, and impaired gag reflex increase the risk of pulmonary aspiration.
(3) Metoclopramide and other antidopaminergic drugs should be avoided, as they may exacerbate preexisting movement disorders.
(4) Adequate hydration and glucose supplementation must be maintained perioperatively.
(5) Metabolic acidosis should be treated with intravenous glucose, lipids, and bicarbonate hydration.
(6) Carnitine deficiency is common and intravenous L-carnitine should be supplemented at double the chronic maintenance dose for surgery (22).
n. Beta-ketothiolase deficiency is a disorder of isoleucine metabolism that presents with intermittent vomiting and acidosis.
(1) Headaches and ataxia are common symptoms.
(2) Infections and protein intake can precipitate acute acidosis. It is unclear if surgery is also a trigger (1).
(3) Serum salicylate levels may be falsely elevated due to reagent cross-reactivity (1).
DISORDER: Urea cycle (see Fig. 31.1)
1. Metabolic disorders of the urea cycle are characterized by hyperammonemia, respiratory alkalosis, and varied neurologic symptoms including encephalopathy or seizures (1).
a. Muscle weakness, vomiting, and headaches associated with cerebral edema may be prominent features.
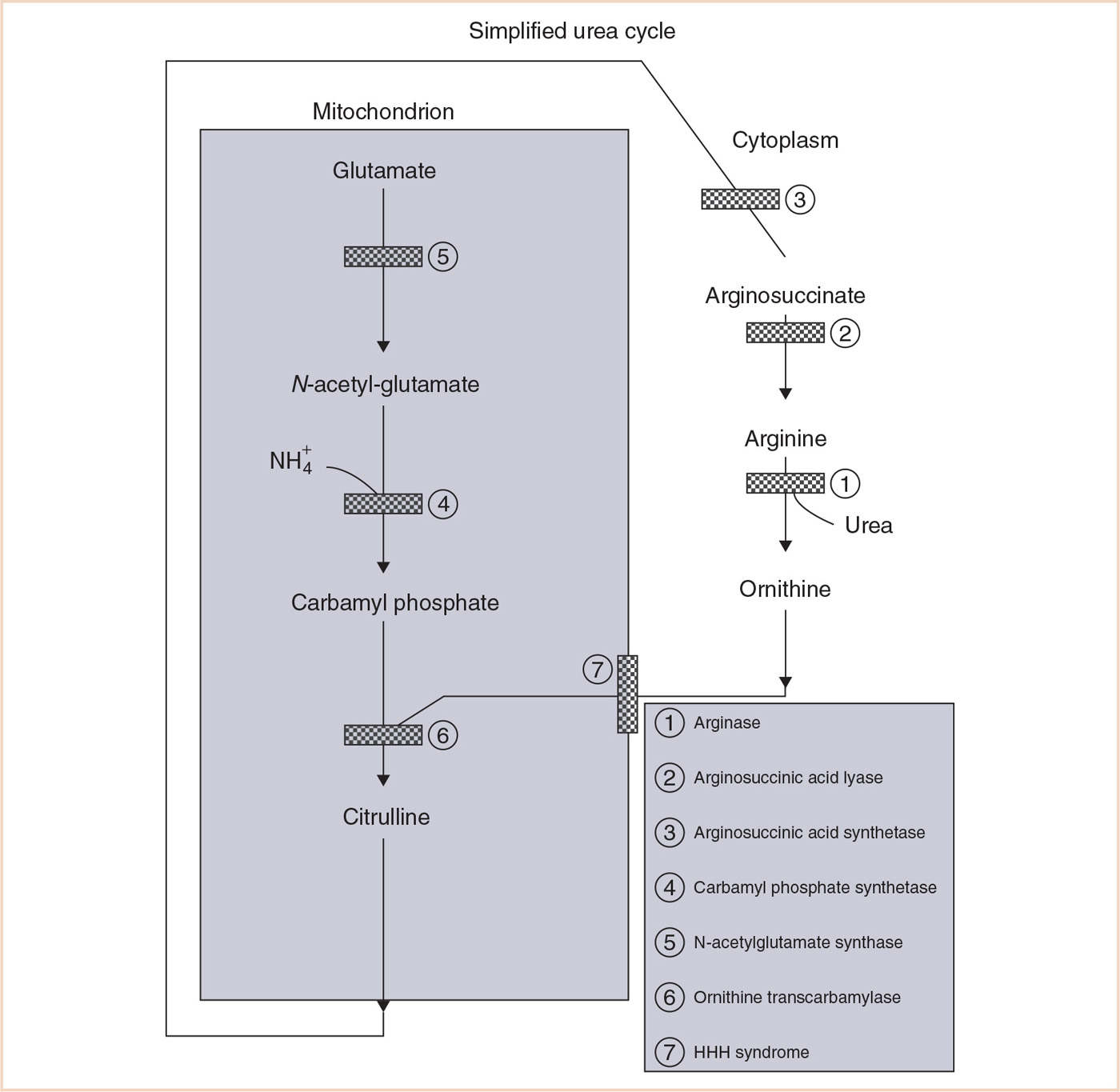
FIGURE 31.1 Simplified urea cycle. (Reproduced with permission from Baum VC, O’Flaherty JE. Anesthesia for Genetic, Metabolic, and Dysmorphic Syndromes of Childhood. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007.)
b. Acute metabolic encephalopathy or ataxia may be precipitated by anesthesia (1).
c. Acid–base status and ammonia levels should be evaluated pre- and postoperatively.
d. Fasting should be minimized and intravenous maintenance fluids containing dextrose initiated to avoid hypoglycemia and prevent catabolism.
e. Gastrointestinal absorption of blood may cause acute decompensation and must be minimized.
f. Encephalopathy should be managed with conservative intravenous hydration (overaggressive use may exacerbate cerebral edema), and lipid and carbohydrate infusion.
g. Seizures are common, but typically easily controlled with phenobarbital or carbamazepine. Valproic acid is contraindicated, as it exacerbates the hyperammonemia (3).
h. Hyperammonemia should be treated with the intravenous nitrogen scavengers sodium benzoate or sodium phenylacetate. Arginine infusions have also been used successfully. Dialysis may be necessary for severe hyperammonemia (3).
i. Supplementation with L-carnitine is common.
j. Parenteral albumin and blood product transfusions have been used safely, but can potentially increase the nitrogen load and precipitate symptoms (23).
k. Intravenous steroids should be avoided, as they increase catabolism (23).
CLINICAL PEARL Metabolic disorders of the urea cycle are characterized by hyperammonemia, respiratory alkalosis, and varied neurologic symptoms including encephalopathy or seizures (1).
2. Carbamoyl phosphate synthetase I deficiency, Citrullinemia, Arginosuccinic acid lyase deficiency (ASLD), and Ornithine transcarbamylase (OTC) deficiency have similar clinical presentations.
a. Hepatic failure may occur in OTC deficiency, the most common of the urea cycle defects.
b. Volatile anesthetics, propofol, opioids, aminosteroid, and tetrahydroisoquinolinium muscle relaxants have been used safely in OTC deficiency (23,24).
c. Patients with ASLD have an increased incidence of hepatic disease and systemic hypertension (25).
3. Arginase deficiency is a urea cycle defect that presents between 1 and 3 years of age with primarily neurologic manifestations (1).
a. Spastic tetraplegia and severe intellectual disability are common with untreated disease.
b. Arginine stimulates nitric oxide production, and patients may be more prone to vasodilation and hypotension with intravenous induction and inhalational anesthetics (26).
c. Case reports suggest increased sensitivity to anesthetic agents (26).
4. Hyperornithinemia–hyperammonemia–homocitrullinuria (HHH) syndrome is a disorder of ornithine transport that presents similarly to urea cycle defects.
a. Confusion, ataxia, and choreoathetosis may be present.
b. Seizures are common and spastic paraplegia has been described.
c. Chronic liver dysfunction with mild coagulopathy may be present (3).
d. Propofol should be avoided because of concerns about increased lipoprotein synthesis and protein metabolism (27).
DISORDER: Carbohydrate metabolism
1. Fructose 1,6 diphosphatase deficiency is a disorder of gluconeogenesis leading to metabolic acidosis.
a. Hypoglycemia complicated by seizures and hypotonia may be present.
b. Perioperative intravenous fluids should contain glucose and ketoacidosis will occur with infection, stress or prolonged fasting.
c. Oral medications formulated in fructose, sucrose, or sorbitol must be avoided (1).
2. Galactosemia is a disorder of galactose metabolism that manifests as seizures, cataracts, and hepatic dysfunction.
a. Hepatomegaly, neonatal hepatic failure, and cirrhosis are common systemic complications (1).
b. Glucose, electrolytes, and liver enzymes should be evaluated preoperatively and followed perioperatively.
c. 10% of patients will have coagulation disorders (3).
d. Epileptogenic sedative and anesthetic drugs should be avoided.
e. Cow’s milk and medications formulated with whey, lactose, or galactose must be avoided (3).
3. Glucose-6-phosphate dehydrogenase (G6PD) deficiency is a hemolytic disorder caused by insufficient erythrocyte-reducing agents.
a. Sulfonamides, hydralazine, nitrates, and methylene blue can trigger acute hemolysis.
b. Preoperative blood cell count and serum bilirubin levels are recommended.
c. Hemolysis typically occurs between 24 and 72 hours after the offending agent is introduced (28).
d. Eutectic mixture of local anesthetic (EMLA) is one potential trigger and should likely be avoided (although there have been no reported complications).
e. Cholelithiasis and reversible renal insufficiency are common (1).
f. Generally, all anesthetic agents (except some local anesthetics) are safe in G6PD deficiency (29) (Fig. 31.2).
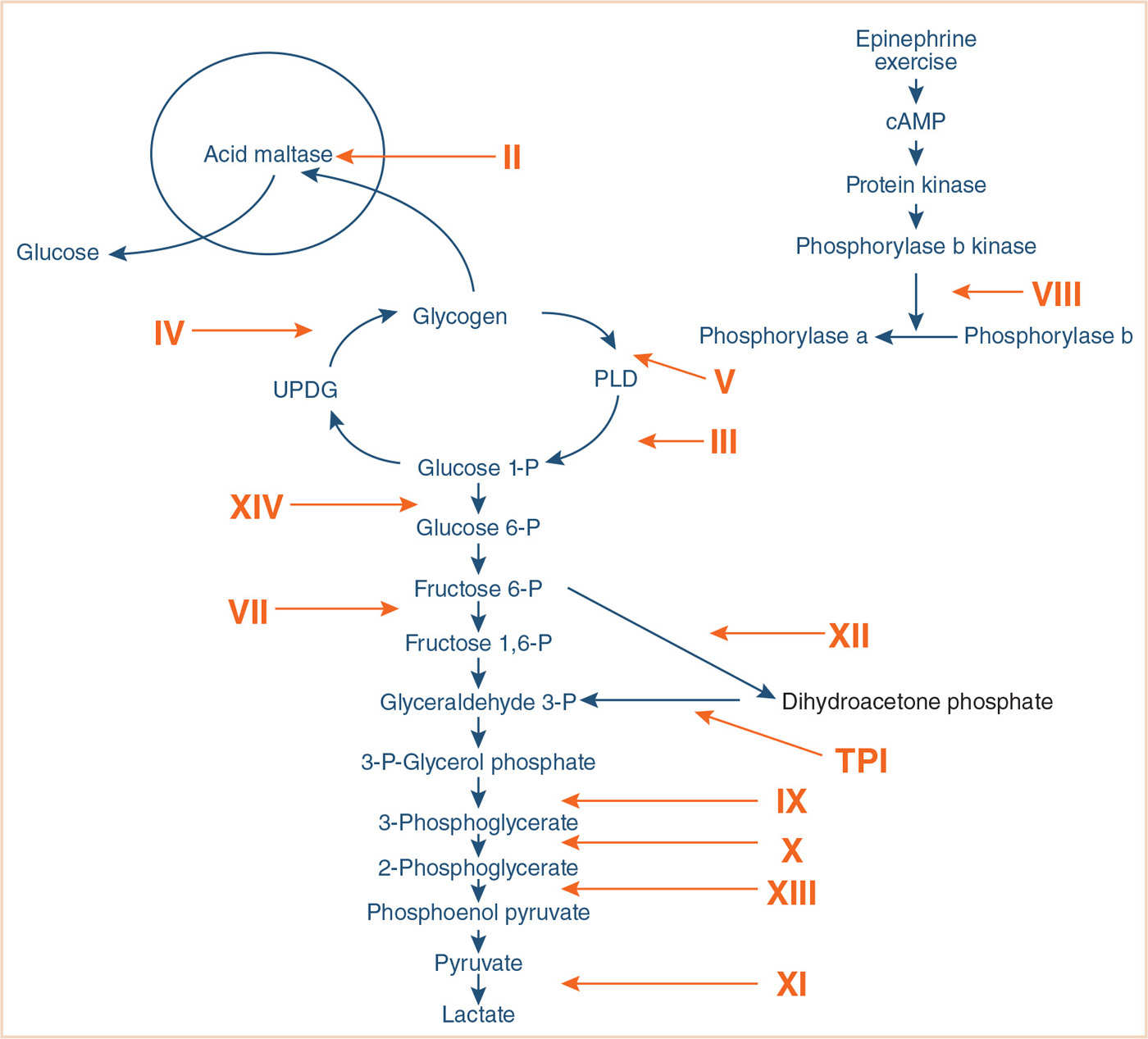
FIGURE 31.2 Glycolytic pathways. Glycogen storage diseases (GSDs) are designated with Roman numerals. GSD I and VI are not included since they do not cause muscle disease. The numerals denote defects in the following enzymes: II, acid maltase; III, debrancher; IV, brancher; V, myophosphorylase; VII, phosphofructokinase; VIII, phosphorylase b kinase; IX, phosphoglycerate kinase; X, phosphoglycerate mutase; XI, lactate dehydrogenase; XII, aldolase A; XIII, beta enolase; XIV, phosphoglucomutase 1. cAMP, cyclic adenosine monophosphate; UDPG, uridine diphosphoglucose; PLD, phosphorylase-limit dextrin; TPI, triose phosphate isomerase. (Reproduced with permission from Tobon A. Metabolic myopathies. Continuum. 2013;19:1571.)
CLINICAL PEARL Glucose-6-phosphate dehydrogenase (G6PD) deficiency is a hemolytic disorder caused by insufficient erythrocyte-reducing agents. Sulfonamides, hydralazine, nitrates, and methylene blue can trigger acute hemolysis.
4. The glycogen storage diseases are a constellation of at least nine different enzymatic disorders of glycogen synthesis, metabolism, or mobilization.
a. Various organ systems are involved in the disorders, including muscle, brain, heart, and liver.
b. Intravenous glucose supplementation while fasting is imperative for nearly all subtypes, and serum glucose must be monitored perioperatively (1).
c. Arterial line placement should be considered in order to monitor acid–base and glucose levels during major surgery.
5. Glycogen storage disease type 0 is a disease of decreased glycogen stores and is characterized by hypoglycemia and seizures.
a. Case reports indicate that hypertrophic cardiomyopathy and sudden death may be associated with this disease process (30).
b. A preoperative electrocardiogram and possibly echocardiogram are recommended.
6. Glycogen storage disease type I (von Gierke disease) is characterized by hypoglycemia, hyperlipidemia, hyperuricemia, and neutropenia (3).
a. Common associated anomalies include massive hepatomegaly, systemic hypertension in adulthood, and platelet dysfunction (1).
a. Platelet function may improve with desmopressin (DDAVP) (1).
b. Renal function should be evaluated preoperatively.
c. Prolonged propofol use is discouraged because of the increased risk for pancreatitis and hyperuricemia (31).
CLINICAL PEARL Prolonged propofol use is discouraged because of the increased risk for pancreatitis and hyperuricemia (31).
7. Glycogen storage disease type II (Pompe disease) is characterized by macroglossia and cardiomyopathy.
a. Treatment with enzyme therapy (alglucosidase alfa) in infancy improves mortality and morbidity (including ventilator-independence and cardiac mass) (3).
b. A critical preoperative airway examination is essential in determining the airway management of these patients. Parents should be queried regarding patient snoring or sleep apnea. Respiratory insufficiency is present in a majority of cases (3).
c. Preoperative electrocardiogram and echocardiogram are necessary to assess ventricular function and the degree of left ventricular outflow tract obstruction.
d. Myocardial depressant drugs and hypovolemia are poorly tolerated secondary to elevated left ventricular filling pressures.
e. Inotropes and afterload-reducing agents may worsen left ventricular outflow tract obstruction.
f. Hypotonia and dysphagia are common and increase the risk of pulmonary aspiration.
g. Succinylcholine is contraindicated as it may cause exaggerated hyperkalemia (1).
h. Hypoglycemia is not characteristic of this disorder.
i. Postoperative intensive care should be arranged to provide respiratory and hemodynamic monitoring.
CLINICAL PEARL Preoperative electrocardiogram and echocardiogram are necessary to assess ventricular function and the degree of left ventricular outflow tract obstruction.
8. Glycogen storage disease type III (Cori disease) is characterized by ketotic hypoglycemia, hepatomegaly, and cardiomyopathy.
a. Massive hepatomegaly and macroglossia are typically present.
b. Hypertrophic cardiomyopathy develops in a majority of cases with the IIIa subtype, and a preoperative electrocardiogram and echocardiogram are indicated (3).
c. Myopathy and osteoporosis are slowly progressive.
d. Coagulopathy is proportional to the degree of liver dysfunction.
e. Intravenous steroids are relatively contraindicated.
f. Preoperative admission is recommended with administration of nocturnal enteral cornstarch and initiation of dextrose-containing intravenous maintenance fluids (3).
9. Glycogen storage disease type IV (Brancher deficiency) is an uncommon form characterized by early cirrhosis.
a. Portal hypertension, coagulopathy, and hypoalbuminemia may result.
b. Preoperative electrolytes, PT/PTT, and a cell blood count should be evaluated.
c. Dilated cardiomyopathy has been described, and a preoperative electrocardiogram and echocardiogram should be considered.
d. Hypotonia may contraindicate the use of succinylcholine.
10. Glycogen storage disease type V (McArdle syndrome) commonly presents as myopathy without hypoglycemia or hepatomegaly.
a. Glucose supplementation may decrease fatty acid metabolism and improve symptoms, but it is not essential.
b. Succinylcholine is contraindicated, and prolonged tourniquets or Bier blocks should be avoided because of the myopathy. Prevention of shivering is important.
c. Obstructive cardiomyopathy and conduction block are rare, but have been described in case reports (3).
d. There is no conclusive association with malignant hyperthermia, but inhalational anesthetics may contribute to rhabdomyolysis; therefore, nontriggering anesthetics have been recommended by many authors (32).
e. Although 50% of individuals have recurrent myoglobinuria, renal failure is rare (3).
f. Intravenous steroids should not be routinely used.
CLINICAL PEARL There is no conclusive association with malignant hyperthermia, but inhalational anesthetics may contribute to rhabdomyolysis; therefore, nontriggering anesthetics have been recommended by many authors (32).
11. Glycogen storage disease type VI (Hers disease) is a milder disease, complicated by hypoglycemia and hepatomegaly.
a. Hypotonia may be present, and succinylcholine should not be used.
b. Cirrhosis may develop slowly (1).
c. Glucagon should not be used to treat hypoglycemia (3).
12. Glycogen storage disease type VII (Tarui disease) presents with muscle weakness and hemolytic anemia.
a. Anemia should be evaluated preoperatively.
b. Arthrogryposis may be present, and intravenous access and positioning may be difficult (1).
c. Cardiomyopathy occurs in the infantile form.
d. No associated hypoglycemia. Intravenous glucose may paradoxically exacerbate symptoms.
e. Tourniquets should be avoided, as they may exacerbate myalgias or cramping.
f. Succinylcholine is relatively contraindicated.
g. Neurologic manifestations may include seizures.
13. Glycogen storage disease type VIII (Phosphorylase kinase deficiency) may present with hepatomegaly large enough to compromise functional residual capacity.
a. Hyperlipidemia and hypoglycemia may be present.
b. Patients may have hypotonia, but there is no associated myopathy (1).
14. Pyruvate dehydrogenase complex deficiency is a mitochondrial defect that presents with metabolic acidosis and ataxia.
a. Prolonged fasting is poorly tolerated and will precipitate neurologic symptoms, including coma.
b. Microcephaly and cleft palate may be associated.
c. Laryngeal stridor has been described with one subtype (1).
d. Hypothermia may precipitate lactic acidosis.
e. Lactated Ringer solution may worsen the lactate burden in these patients.
f. Respiratory depression is common, and patients should be monitored postoperatively (1).
15. Pyruvate carboxylase deficiency is a mitochondrial disorder characterized by developmental delay, seizures, and metabolic acidosis (3).
a. Fasting patients should be maintained on a lactate-free, dextrose-containing intravenous solution.
b. Most forms have severe neurologic complications and are lethal in infancy.
c. Citrate and aspartic acid supplementation provide alternative energy substrates (3).
d. Patients are at risk for central respiratory depression and postoperative apnea.
e. The use of propofol remains controversial. To date, there are no case reports of propofol use in this disorder.
DISORDER: Fatty acid metabolism
1. Abetalipoproteinemia is a rare disorder of lipoprotein metabolism that can lead to skeletal myopathy, peripheral neuropathy, and ataxia.
a. Coagulopathy may result from malabsorption of vitamin K (1). Anemia has been described.
b. Spinocerebellar degeneration leading to hypotonia contraindicates the use of succinylcholine.
c. Regional anesthesia may be complicated by the suggestion of decreased efficacy of local anesthetics (1).
d. Cardiomyopathy has been described in a case report.
2. Glutaric aciduria II (Multiple acyl-coenzyme A (CoA) dehydrogenase deficiency) is a disorder of fatty acid oxidation that results in an organic aciduria and nonketotic acidosis.
a. Respiratory distress and pulmonary hypoplasia are described.
b. Hypotonia and regular vomiting increase the risk of pulmonary aspiration.
c. Patients are at risk for pulmonary arterial hypertension and hypertrophic cardiomyopathy; therefore, a preoperative echocardiogram should be considered (1).
d. Hepatomegaly and renal dysplasia warrant preoperative laboratory evaluation.
e. Fasting should be minimized and nonlactated, dextrose-containing maintenance intravenous fluids started preoperatively.
f. Nitroprusside should be used with caution as it inhibits electron transport (1).
g. Propofol, especially as an infusion, should probably be avoided.
3. Short-chain acyl-coenzyme A dehydrogenase deficiency (SCAD) differs from other disorders of fatty acid oxidation in that it rarely presents with severe hypoglycemic episodes.
a. Infants may have seizures, hypotonia, and developmental delay (3).
b. Many individuals with SCAD are asymptomatic (3).
c. Hypoglycemia and metabolic acidosis may only be evident with infection or fasting.
d. Avoid fasting longer than 12 hours (3). Glucose should be supplemented at a rate of 6 to 10 mg glucose/kg/minute (3).
e. Lactated Ringer solution is not contraindicated in this disorder (1).
f. Succinylcholine should be avoided in patients with associated myopathy.
g. Propofol contains predominantly long-chain fatty acids, which are metabolized normally in this disorder.
4. Medium-chain acyl-coenzyme A dehydrogenase deficiency (MCAD) is the most common fatty acid oxidation disorder.
a. Patients may present with intermittent hypoglycemia, hyperammonemia, and metabolic acidosis (1).
b. Seizures, coma, and sudden death can be associated with an acute illness (3).
c. Secondary carnitine deficiency may not be beneficial, and can be detrimental (1).
d. Perioperative glucose supplementation is essential (10 to 12 mg glucose/kg/minute) (3).
e. Fasting should be limited to 8 hours (infants 6 to 12 months), 10 hours (infants 12 to 24 months), and 12 hours (infants older than 24 months) (3).
f. Hepatomegaly and acute liver dysfunction may be the presenting signs (3).
g. Symptomatic patients merit preoperative arterial blood gas, cell blood count, electrolytes, liver function tests, and ammonia and serum lactate levels (3).
h. Propofol infusions should likely be avoided, although single induction doses are probably tolerated.
CLINICAL PEARL Medium-chain acyl-coenzyme A dehydrogenase deficiency (MCAD) is the most common fatty acid oxidation disorder. Patients may present with intermittent hypoglycemia, hyperammonemia, and metabolic acidosis (1).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


