KEY POINTS
Acute mesenteric ischemia is an infrequent but deadly clinical entity. When diagnosis is delayed, it is almost always fatal; therefore, a high index of suspicion is required, especially in those at high risk: the elderly, those with cardiac dysfunction, patients with diffuse atherosclerosis, and those following aortic and cardiac surgery or arterial catheterization.
The etiology of acute mesenteric ischemia may be embolic, thrombotic, primary vasoconstrictive, or secondary to venous thrombosis. Chronic ischemia is usually due to flow-limiting lesions (mesenteric stenosis or occlusions) in the presence of inadequate collateralization.
Classic symptoms of acute intestinal ischemia are central abdominal pain (often out of proportion to the benign abdominal examination), weight loss (an important clue even in the acute presentation), bowel emptying, and altered bowel function (vomiting, bloating, constipation, or diarrhea). Once signs of peritonitis or bloody diarrhea are present, shock, sepsis, and death almost always follow.
Arterial phase abdominal and pelvic computed tomographic (CT) mesenteric angiography is the investigation of choice, offering accurate diagnostic evaluation. However, selective mesenteric angiography offers therapeutic options, whereas duplex ultrasonography may not be definitive. Frequently, the diagnosis is confirmed only at laparotomy.
Treatment is most commonly surgical, with restoration of flow by embolectomy, bypass, or angioplasty (antegrade or retrograde); vasodilator infusion therapy; thrombolysis and resection of nonviable intestine; and liberal use of “second look” laparotomy.
Nonocclusive mesenteric ischemia (NOMI) has a high mortality rate, and early diagnosis and treatment are important for improving survival in patients with this condition.
Acute mesenteric ischemia is a relatively rare but often fatal clinical entity. Although little data exist on its true incidence, data from the Swedish Vascular Registry suggest that it may account for just 1% of reconstructions for acute thromboembolism.1 Contemporary series, however, continue to report a mortality rate of between 32% and 48%.2,3 Although autopsy studies suggest that atherosclerosis affecting the mesenteric arteries is common (6%-10%),4 symptomatic mesenteric occlusive disease is rare. However, of patients presenting with acute mesenteric ischemia, one large series found that 43% had prior symptoms of chronic mesenteric ischemia.5 The spectrum of mesenteric ischemia includes occlusive disease secondary to atherosclerotic occlusion with thrombosis, embolism, mesenteric venous thrombosis, and nonocclusive mesenteric ischemia due to vasospasm (Table 109-1). At its most florid, it may present with mesenteric infarction, intestinal perforation, and septic circulatory collapse. This relatively rare but often fatal clinical entity must be considered early in the differential diagnosis of any patient with abdominal symptoms or signs but especially those with pain out of proportion to physical findings. Also a history of intestinal angina, peripheral vascular disease, cardiac dysfunction, aortic surgery or recent aortic catheterization, hypotension, or prothrombotic state increases the risk of mesenteric vascular disease. Noninvasive tests for mesenteric ischemia lack specificity and sensitivity, which mandates that the diagnosis often requires a high index of suspicion, supplemented by a liberal use of computed tomographic angiogram (CTA) when uncertainty remains. Where doubt exists in the presence of emerging acute abdominal signs or clinical deterioration, diagnostic laparotomy is indicated. This discussion will focus on the etiology, pathophysiology, diagnosis, and management of acute mesenteric ischemia.
Potential Causes of Acute Mesenteric Ischemia
| Occlusive Disease | |
| Embolism | Cardiac diseases (atrial fibrillation, post-MI, valvular disease, |
| SBE, dilated left ventricle, myxoma) | |
| Extracardiac arterial diseases | |
| Arterial thrombosis | Acute on chronic atherosclerosis |
| Low cardiac output states | |
| Intrinsic arterial diseases | Occlusive atherosclerosis |
| Aortic dissection (type A or B) | |
| Atherosclerotic aneurysm | |
| Arteritis and autoimmune diseases | |
| Fibromuscular dysplasia | |
| Venous thrombosis | Thrombophilia |
| Extrinsic compression | |
| Iatrogenic | After aortoiliac surgery |
| Catheter related (dissection, embolism) | |
| Irradiation arteritis | |
| Vasoconstrictive agents (epinephrine, norepinephrine, dopamine) | |
| Trauma | Penetrating |
| Blunt (including deceleration injuries) | |
| Nonocclusive Disease | |
| Shock | Cardiogenic shock |
| Hypovolemic shock | |
| Septic shock | |
| Neurogenic shock | |
| Anaphylactic shock | |
| Low cardiac output states | Heart failure |
| Arrhythmia | |
| Acute coronary syndromes | |
| Miscellaneous : peritonitis, pancreatitis, post CABG, ESRD on peritoneal dialysis | |
| Pseudocoarctation (aortic dissection) | |
| Pharmacologically induced | Digitalis, vasoactive substances (catecholamines, somatostatin analogues, etc.), ergotism |
ANATOMY AND DYNAMICS OF THE MESENTERIC CIRCULATION
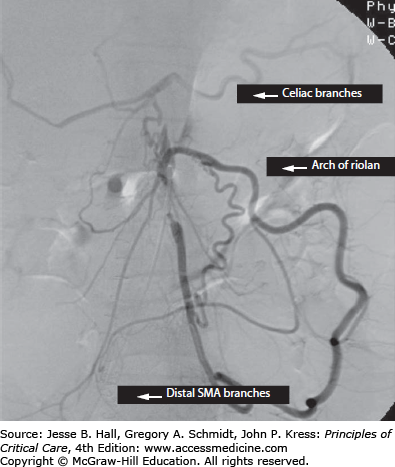
The mesenteric circulation is supplied in series by the three major midline branches of the abdominal aorta, namely the celiac artery, supplying the foregut, hepatic, and splenic beds; the superior mesenteric artery (SMA), supplying the midgut; and the inferior mesenteric artery (IMA) and the internal iliac arteries, supplying the hindgut. An extensive network of actual and potential sites of collateralization exists between individual branch territories and their neighbors, as well as the systemic circulation. The celiac territory may gain supply proximally across the diaphragm from the phrenic and esophageal vessels (that arise from the aorta) and distally from the SMA via the gastroduodenal artery, including the superior and inferior gastroduodenal arteries. The SMA territory may be perfused from the celiac artery, as mentioned, or from the IMA territory via the arch of Riolan, a collateral that runs in the midmesentery and is fed via the ascending branch of the left colic artery (Fig. 109-1). The IMA territory may collateralize proximally, as described, or distally via the inferior hemorrhoidal arteries from the internal iliac artery. Blood from the pelvis may collateralize to the SMA or above from the communications between the sigmoidal/hemorrhoidal vessels to the marginal artery of Drummond (a collateral that runs within 1-2 cm of the mesenteric edge of the bowel) and via the arch of Riolan. Vessel caliber decreases progressively from the main aortic branch to the mesenteric vascular arcades distally, ending with an extensive communicating submucosal vascular plexus. This collateral network explains why many individuals may tolerate chronic occlusion of one or two mesenteric vessels without symptoms, but it also explains why acute-on-chronic occlusion of one additional branch may lead to a catastrophic loss of intestinal perfusion.
CONTROL OF THE MESENTERIC CIRCULATION
The mesenteric circulation receives approximately 20% to 30% of the cardiac output at rest, which may increase by up to 50% after meals.6 As such, the mesenteric circulation receives approximately three times more blood per unit weight than most other body tissues. This blood flow is partly to satisfy the absorptive function of the intestine and perfuse the liver via the portal vein, but it also represents a reservoir function from which blood can be mobilized to other sites (vital organs) at times of stress or increased demand. This preferential shunting of blood to vital organs, if acute or severe, can “sacrifice” the mesenteric circulation, leading to low-flow and ischemic injury. The more metabolically active mucosal layer receives 70% of blood flow, only 30% supplying the muscularis and serosal layers, placing the intestinal mucosa at greatest risk from ischemic injury.6 Within the intestinal villus, passive exchange of oxygen typically occurs between the afferent arteriole and efferent venule, effectively bypassing the capillary network at the villus tip, a phenomenon called oxygen countercurrent exchange. In health, high partial pressures of oxygen ensure that the metabolic needs of the villus mucosa are met despite this shunting, but in deoxygenated states this shunting adversely affects oxygen delivery to the mucosal tip, making it most vulnerable to ischemic injury. A number of extrinsic and intrinsic factors regulate the mesenteric blood flow, leading to a complex interaction between neural, hormonal, and paracrine effectors that regulate the vascular smooth muscle tone in the mesenteric bed and control local blood flow. Vasoactive mediators alter the vascular smooth muscle (VSM) tone of multiple small afferent arterioles, collectively known as resistance vessels, changing their cross-sectional area and blood flow (Table 109-2). The interaction between extracellular agonist (first messenger) and VSM receptor leads to accumulation of intracellular second messengers, such as cyclic adenosine monophosphate (cAMP), Ca2+, and cyclic guanosine monophosphate (cGMP). These second messengers directly or indirectly alter the cytosolic concentration of Ca2+ and dictate whether VSM contracts.7 A functioning cardiovascular system is essential because the mesenteric circulation is often sacrificed to maintain blood flow to vital organs at times of detrimental alterations in cardiac output, blood volume, or arterial blood pressure. Increased sympathetic nervous activity associated with cardiogenic, septic, or hypovolemic shock can further compound flow-related ischemia by inducing intense vasoconstriction within the mesenteric bed. High-volume hemorrhage (>35% of blood volume) leads to disproportionate visceral vasoconstriction compared with the reduction in cardiac output.8 When activated postganglionic sympathetic nerves fibers release norepinephrine, stimulating α2- adrenergic receptors on VSM of precapillary arterioles (which are the resistance vessels), this results in vasoconstriction and reduced intestinal blood flow. Some compensation is provided by the simultaneous stimulation of vasodilatory β2-adrenergic receptors on VSM by norepinephrine.9 However, the net effect is a rapid reduction in flow followed by a gradual return to prestimulation blood flow levels, a phenomenon known as autoregulatory escape. Exogenously administered norepinephrine similarly can cause nonocclusive mesenteric ischemia (NOMI). Parasympathetic nerves induce vasodilation due to the effect of specific neurotransmitters acetylcholine (ACh, increases cGMP and NO), vasoactive intestinal peptide (VIP, increases cAMP), and adenosine triphosphate (ATP). Primary sensory nerves also play a role because their activity through C fibers can inhibit sympathetic impulse flow through the spinal cord or sympathetic ganglia. Indeed, direct antidromic vasodilation can occur as C fibers release neuropeptides, including substance P, calcitonin gene–related peptide, and VIP, in response to luminal signals. Humoral (endocrine) messengers also can affect the mesenteric blood flow, most notably adrenal catecholamines in states of systemic stress, shock, or secreting tumor (pheochromocytoma). Similarly vasoconstrictive effects are seen with renal-derived angiotensin II in congestive heart failure and pituitary-derived vasopressin in shock, respectively. Ingested meals and gastrointestinal luminal peptides stimulate a postprandial increase in mesenteric blood flow. This vasodilatory response is regulated, in part, by the increased metabolic activity in the tissues, leading to local accumulation of vasoactive metabolites such as amines, peptides, prostanoids, and adenosine in the presence of a reduced oxygen concentration. Adenosine itself, increasing as a result of energy-dependent processes, may vasodilate in a paracrine fashion. A number of hormones are also released in response to luminal contents, including gastrin, cholecystokinin, and secretin, with net vasodilatory effects.10 Somatostatin, released by the mucosal D cells, induces mucosal vasoconstriction, and synthetic analogs (eg, octreotide) can induce ischemia.
Potential Vasoactive Mediators of the Enteric Circulation
| Mediator | Vasoconstrictors | Vasodilators |
|---|---|---|
| Neural | ↑ Sympathetic tone (adrenergic) | ↓ Sympathetic tone (cholinergic) |
| ↓ Parasympathetic tone | ↑ Parasympathetic tone | |
| Neuropeptide Y | Substance P | |
| Vasoactive intestinal peptide (VIP) | ||
| Calcitonin gene-related peptide (CGRP-α) | ||
| Humoral | Catecholamines (except liver and muscle) | Catecholamines (only liver and muscle) |
| Angiotensin II | Histamine | |
| Vasopressin | Bradykinin | |
| Serotonin | Activated complement (C3a, C5a) | |
| Activated complement (C5a) | Adrenomedullin | |
| Paracrine/autocrine | Endothelin-1 (VSM cells) | Endothelium-derived relaxing factor (EDRF) |
| Platelet-activating factor | ||
| Constrictor prostaglandins (F2α) | Endothelium-derived hyperpolarizing factor | |
| Dilator prostaglandins (I2 or prostacyclin) | ||
| Endothelin-1 (endothelial cells) | ||
| Metabolic | ↑ <SPAN role=presentation tabIndex=0 id=MathJax-Element-1-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PO2′>[Math Processing Error]PO2 | ↓ <SPAN role=presentation tabIndex=0 id=MathJax-Element-2-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PO2′>[Math Processing Error]PO2 |
| ↓ <SPAN role=presentation tabIndex=0 id=MathJax-Element-3-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PCO2′>[Math Processing Error]PCO2 | ↑ <SPAN role=presentation tabIndex=0 id=MathJax-Element-4-Frame class=MathJax_Error style="POSITION: relative" data-mathml='PCO2′>[Math Processing Error]PCO2 | |
| ↑ pH | ↓ pH | |
| ↓ Metabolites (K+, lactate, adenosine, etc) | ↑ Metabolites |
PATHOPHYSIOLOGY OF INTESTINAL ISCHEMIA
The unique arrangement whereby the mesenteric blood flow continues in series via the portal vein, supplying much of the metabolic need of the liver, means that oxygen extraction by intestinal tissue remains low at rest. Therefore, intestinal tissues may respond to reduced flow by increasing oxygen extraction from the blood without a requirement for increased blood flow, but this deprives the liver of some of its portal-derived oxygenation, resulting in hepatic ischemia. On a biochemical and microscopic level, acute ischemia of the intestine is characterized by depletion of cellular adenosine triphosphate (ATP). This leads to failure of the key energy-dependent processes in the cells (in particular membrane ATPase pumps) and results in cellular swelling, membrane disruption, metabolic arrest, and cell death.11 Hypoxic conditions themselves favor the conversion of the abundant amounts of the enzyme xanthine dehydrogenase to xanthine oxidase. Hypoxia results in accumulating adenosine levels which can be metabolized further to inosine and then hypoxanthine but no further without oxygen. With the reintroduction of oxygen on reperfusion, xanthine oxidase converts hypoxanthine into xanthine, producing superoxide (reperfusion tissue injury12). Oxygen free-radicals (OFRs), including superoxide anions (O2−), hydrogen peroxide (<SPAN role=presentation tabIndex=0 id=MathJax-Element-5-Frame class=MathJax_Error style="POSITION: relative" data-mathml='H2O2′>[Math Processing Error]H2O2), and hydroxyl radicals (OH−), arise as by-products of the xanthine-xanthine oxidase system, the mitochondrial electron transport system, and the NADPH oxidase system of infiltrating neutrophils. Endogenous defenses against oxidative injury, including intracellular enzymes (superoxide dismutase, catalase, and glutathione peroxidase) and vitamins (C and E), can be overwhelmed by the production of OFRs during reperfusion. These OFRs directly attack cell membranes, resulting in lipid peroxidation and synthesis of lipid mediators (thromboxane A2, leukotriene B4) and chemotactic peptides (complement component C5a), which promote neutrophil activation and chemotaxis, augmenting the inflammatory component. Sequestration of circulating neutrophils in the microvessels of the intestine begins with constitutively expressed neutrophil L-selectin receptors binding to the postcapillary venule endothelial P-selectin and E-selectin receptors.13 This is the first phase of neutrophil adhesion, manifested by neutrophil rolling along the venular luminal surface. A second phase of neutrophil adhesion and influx into the tissue occurs after upregulation of the CD11/CD18 integrins and shedding of L-selectin from the neutrophil surface, caused by release of proinflammatory cytokines, platelet-activating factor (PAF), and eicosanoids from the damaged endothelial cell. This halts neutrophil rolling in the blood vessels of the intestine and by providing strong neutrophil-endothelial cell adhesion through the interaction of the CD11b/CD18 integrin with the endothelial receptor, intercellular adhesion molecule 1 (ICAM-1).14 Finally, neutrophils transmigrate in response to chemotactic stimuli (such as interleukin-8) and binding by platelet-endothelial cell adhesion molecule 1 (PECAM-1). Neutrophils further damage tissues by releasing superoxide anions via the NADPH oxidase system, secreting myeloperoxidase that catalyzes the production of hypochlorous acid (HOCL), and releasing granular enzymes, including elastase, collagenase, and cathepsin G.
In addition, many capillaries fail to perfuse on reinstitution of blood flow, the no-reflow phenomenon, due to loss of local autoregulation combined with mechanical obstruction of narrowed capillaries by enlarged adherent neutrophils.15 No-reflow results in incomplete and patchy tissue reperfusion, prolonging hypoxia and exposure of the tissues to toxic metabolites. On a microscopic level, complete ischemia causes detectable injury to the superficial part of the mucosa (the villi) within 20 minutes. As the duration of ischemia increases, the villus loss can be complete before the muscular layers are damaged. Patchy cellular necrosis can extend to mucosal sloughing seen clinically as bloody diarrhea. Ultimately, there is transmural infarction, perforation, and septic peritonitis. Prior to reperfusion, metabolic products such as lactate and other acids are not absorbed via the negligible portal circulation but only through the peritoneal surfaces, accounting for the lack of acidosis or elevated serum lactate commonly observed in patients presenting with acute mesenteric ischemia.
SYSTEMIC RESPONSE TO MESENTERIC REVASCULARISATION
On reperfusion of the acutely ischemic intestine, local injury can induce a systemic response as metabolic byproducts of the ischemic process (lactate, H+, K+, oxygen reactive species, arachidonic acid derivatives, cytokines, possibly endotoxin, and activated leukocytes) are flushed into the portal and systemic circulation. A systemic inflammatory response syndrome (SIRS) is initiated, triggering the complement and coagulation cascades, elaborating cytokines, and provoking widespread endothelial dysfunction and vital organ injury. Acute lung injury (ALI) is the most common manifestation of the systemic inflammatory response to intestinal reperfusion.
Though surviving this initial phase of acute gut-derived inflammation, the reperfused but injured intestine can continue to be detrimental to the host. The gastrointestinal tract normally is inhabited by a large collection of gram-negative bacteria. Gut-mucosal barrier disruption after ischemia-reperfusion injury allows translocation of these bacteria.16,17 The vast hepatic sinusoid network, lined with fixed-tissue macrophages (Kupffer cells), is strategically located to interact with gut-derived endotoxin and contribute to multiple-organ-system failure (MOSF). Noncardiogenic pulmonary edema (ALI) is well recognized after intestinal ischemia reperfusion injury.14,18 Postoperative, renal, and hepatic dysfunctions also are common.
INTESTINAL ISCHEMIA RISK FACTORS
A number of factors influence the development of and survival from acute mesenteric ischemia. Factors predisposing to occlusive mesenteric ischemia include hypertension, tobacco use, family history of cardiovascular disease, peripheral vascular disease, coronary artery disease, diabetes, congestive heart failure, prior myocardial infarction, cerebrovascular disease, hypercholesterolemia, and atrial fibrillation.19,20 Nonocclusive mesenteric ischemia results from splanchnic vasoconstriction in response to systemic factors and is associated with acute cardiac dysfunction (myocardial infarction, arrhythmia, left ventricular dysfunction), hypovolemia, and pharmacotherapy.21 Digitalis is an additional risk factor for developing NOMI. It induces vasoconstriction and thus an increased resistance in peripheral splanchnic vessels.59 Alkaloids constitute another substance group causing smooth muscle contraction of the arteriolar wall. Ergotamine is one of the most potent vasoconstrictors in this group, which plays an important pathogenic role in NOMI.60,61 A combination of glycosides and diuretics is frequently administered to patients with congestive heart failure. The increased renal blood flow caused by furosemide leads to a diminished mesenteric perfusion. This is probably due to the furosemide-related activation of the renin–angiotensin–aldosterone system with subsequently increased levels of angiotensin II.62,63 Other causes of mesenteric vasospasm are various forms of shock, septicemia, dehydration, and hypotension following dialysis and heart surgery or major abdominal surgery.64 The frequent concomitance of pancreatitis in NOMI is explained by the proximity of the superior mesenteric artery (SMA) and the celiac plexus to the pancreas. The inflammation of the pancreas may induce a vasoconstrictive response in the superior mesenteric artery.62
Factors predictive of mortality secondary to intestinal ischemia include advanced age, generally poor health, diagnostic delay, and nonocclusive mesenteric ischemia.5 Most patients with asymptomatic high-grade stenosis or occlusions of all three mesenteric trunks will develop symptoms or die during follow-up.22 Therefore, elective revascularization should be considered if they are fit for surgery.
CLINICAL PRESENTATION OF MESENTERIC ISCHEMIA
The classic diagnostic triad for chronic intestinal ischemia includes weight loss, abdominal angina, and altered bowel habit.4 These patients are frequently investigated for malignancy before chronic mesenteric ischemia is considered. Acute mesenteric ischemia may present on a background of chronic intestinal angina or de novo
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


